

In this issue of Blood, made it their mission to understand the molecular mechanisms by which immunomodulatory drugs (IMiDs) induce thrombocytopenia. The authors use a combination of in vitro and ex vivo methods to show that treatment regimens, including IMiDs in multiple myeloma (MM), lead to aromatase degradation in human megakaryocytes. This has an impact on the estradiol signaling required for proplatelet formation, thus resulting in thrombocytopenia (see figure).1
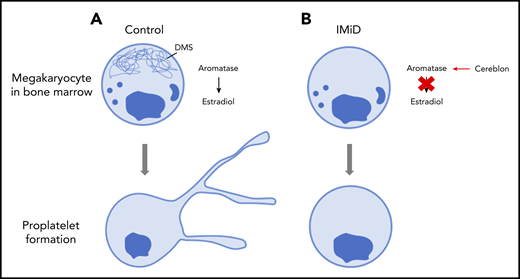
Proplatelet formation. (A) Megakaryocytes generate platelets by remodelling their cytoplasm into long, beaded extensions, called proplatelets, that function as the assembly lines of platelet production. The demarcation membrane system (DMS), an elaborate invaginated membrane network found in mature megakaryocytes, reorganizes to provide the abundant surface membrane material required for proplatelet formation. (B) IMiDs cause aromatase to become a neo-substrate of cereblon preventing endogenous estradiol synthesis and demarcation membrane formation in human megakaryocytes. Lack of demarcation membrane causes abolition of proplatelet formation.
Proplatelet formation. (A) Megakaryocytes generate platelets by remodelling their cytoplasm into long, beaded extensions, called proplatelets, that function as the assembly lines of platelet production. The demarcation membrane system (DMS), an elaborate invaginated membrane network found in mature megakaryocytes, reorganizes to provide the abundant surface membrane material required for proplatelet formation. (B) IMiDs cause aromatase to become a neo-substrate of cereblon preventing endogenous estradiol synthesis and demarcation membrane formation in human megakaryocytes. Lack of demarcation membrane causes abolition of proplatelet formation.
The platelet journey begins in the bone marrow niche where hematopoietic stem cells (HSCs) reside. Some of these HSCs become lineage committed and eventually form mature megakaryocytes. After the maturation process, these megakaryocytes are able to undergo elaborate membrane remodeling and reorganization to extend long protrusions termed proplatelets. These proplatelets function as the assembly lines of platelet production and extend out of the bone marrow and into the sinusoids. Poorly understood fission events within the proplatelet body then lead to the formation of individual blood platelets.2,3 If these fragile, intermediate processes are disturbed, the result may be a reduction in the generation of new platelets and may lead to thrombocytopenia. Other possible explanations for thrombocytopenia include consumption of platelets or the increase in platelet clearance.4
MM is characterized by the dramatic rise in the overall percentage of abnormal plasma cells within the bone marrow population.5 Myelodysplastic syndromes (MDSs) are a group of disorders of the bone marrow typically characterized by cytopenias.6 MDS associated with chromosome 5q31 deletion is of particular interest because it is treated with IMiDs such as lenalidomide. Treatment of MM and MDS is often approached by using a regimen that includes IMiDs such as lenalidomide or pomalidomide, both of which are synthetic chemical modifications of the infamous compound thalidomide.7,8 However, studies have shown that IMiDs may cause the unwanted adverse effects of anemia and neutropenia, because of the importance of the transcription factor Ikaros family zinc finger protein 1 (IKZF1) in erythroid and myeloid differentiation, which has an impact on the treatment of patients with MM.9
To determine what causes IMiD-induced thrombocytopenia, Tochigi and colleagues took a stepwise approach to analyzing the effects of IMiDs on megakaryocyte development and platelet production. The authors first confirmed that there was not a reduced number of megakaryocytes in the marrow and then proceeded to show that IMiDs do not inhibit either maturation or endomitosis of megakaryocytes. If the number of megakaryocytes is still the same and they are at the same level of maturity, then what is causing the thrombocytopenia? IMiDs could potentially affect proplatelet formation. When megakaryocytes were exposed to the lenalidomide or pomalidomide IMiDs, the formation of proplatelets was severely limited, suggesting a significant block in platelet production. This was supported by electron micrographs that showed strikingly reduced demarcation membrane development within the IMiD-exposed megakaryocytes which, under normal circumstances, would provide an elaborate reservoir of redundant membrane important for proplatelet formation.
To identify the molecular mechanisms by which IMiDs were significantly decreasing proplatelet formation, the authors’ first steps were to perform transcriptome analysis of megakaryocytes derived from CD34+ hematopoietic stem/progenitor cells in the presence or absence of the IMiDs. Data analysis showed that the gene expression signatures of estradiol signaling were deficient in the megakaryocytes treated with lenalidomide. This was consistent with previous literature that suggested the estradiol pathway is important for proplatelet formation.10 Also supporting this is the fact that the addition of exogenous estradiol to IMiD-treated megakaryocytes in culture almost completely restored the number of proplatelet-producing megakaryocytes to that of untreated controls. Furthermore, expression of aromatase, an enzyme that mediates estradiol synthesis, was significantly reduced in CD34+-derived human megakaryocyte cultures 24 hours after treatment with IMiDs. Co-immunoprecipitation of lysates of aromatase and cereblon (the direct target of IMiDs) from K562 cells showed that cereblon appeared with aromatase only when lenalidomide was present. Co-immunoprecipitations with a mutant cereblon that was missing the IMiD binding region failed to show binding, as expected. Bone marrow samples of MM patients receiving IMiDs as part of their treatment were compared with those from patients who were not receiving IMiDs. Remarkably, those patients with IMiD-induced thrombocytopenia did not have detectable levels of aromatase within the bone marrow or within isolated megakaryocytes. However, patients not treated with IMiDs had normal levels of aromatase in their bone marrow.
What about the future? The authors have already highlighted the possibility of administering estradiol in the clinic with its implications for thrombosis and postmenopausal breast cancer. The sensible approach would be to understand on a structural basis why the IMiD-cereblon structures are binding to aromatase and then use this knowledge to identify IMiDs that do not facilitate cereblon and aromatase binding. These findings also raise new questions regarding proplatelet formation. It can be appreciated that proplatelets are not typically formed in static conditions in vivo. Instead, they are released by megakaryocytes that are packed in a dense marrow environment and are released with shear forces. It would be interesting to investigate the roles and the importance of aromatase and estradiol in proplatelet formation under physiological shear forces using platelet bioreactors. Is it possible to add increasing concentrations of exogenous estradiol and generate more (pro)platelets? If so, would the platelets that are produced function normally? Would we also be able to inhibit estradiol synthesis and learn about the downstream effects, if any, this inhibition could have on the cytoskeleton arrangement? Tochigi and colleagues have done an excellent job combining their ex vivo and mechanistic data to provide a strong argument that IMiDs cause thrombocytopenia through inhibition of proplatelet formation.
Conflict-of-interest disclosure: J.E.I. is a founder of and has financial interest in Platelet BioGenesis, a company that aims to produce donor-independent human platelets from human-induced pluripotent stem cells at scale. N.L.A. declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal