

TO THE EDITOR:
Follicular T-cell lymphoma (FTCL) is a rare nodal mature T-cell neoplasm included in a broader category of angioimmunoblastic T-cell lymphoma (AITL) and other nodal lymphomas of T follicular helper (TFH) cell origin by the 2017 World Health Organization classification of tumors of hematopoietic and lymphoid tissues.1 The atypical, clear, medium-size neoplastic cells display a common TFH phenotype with expression of CD4, CD10, BCL6, PD-1, CXCL13, and ICOS.2 In contrast to AITL, FTCL is characterized by a follicular growth pattern and lacks the proliferation of high endothelial venules and the extrafollicular expansion of follicular dendritic cells. The molecular pathology of FTCL remains incompletely understood. Up to 40% of FTCLs harbor t(5;9)(q33.3;q22.2) fusing the N-terminal part of the interleukin-2 (IL-2)–inducible T-cell kinase (ITK) to the tyrosine kinase domain of SYK (the spleen tyrosine kinase).2-4 The ITK-SYK fusion protein acts as a constitutively active SYK tyrosine kinase with in vitro and in vivo oncogenic properties.5,6 Notably, mutations in TET2, DNMT3A, and RHOA recurrently occurring in AITL and other TFH-derived lymphomas4,7 were recently identified in a few patients with FTCL whose samples were analyzed,4 underpinning the view that TFH-derived lymphomas represent variants of the same disease.
To obtain additional insight into the molecular pathogenesis of FTCL, we performed cytogenetic and molecular studies of 6 new patients from the University Hospital of Leuven and Université Catholique de Louvain Mont-Godinne, Namur, Belgium. Pathology and clinical records were reviewed. The institutional review board (Commissie Medische Ethiek) of the University Hospital approved this retrospective study and renounced the need for written informed consent (study no. S56035, ML10127: 31/01/2014). The methods used in the study can be found in supplemental Methods (available on the Blood Web site).
All tumors were negative for ITK-SYK, as demonstrated by an initial fluorescence in situ hybridization (FISH) assay (data not shown). Gene expression profiling by RNA sequencing (RNA-seq) showed that results from FTCL patients clustered together, overlapped with peripheral T-cell lymphoma not otherwise specified, and were different compared with normal lymph nodes and anaplastic large cell lymphoma (Figure 1A). Relevant clinical, pathologic, and genetic data from the patients we reported on are shown in Table 1. Notably, all of them were female, although FTCL shows a slight male predominance.2 Their ages ranged from 58 to 83 years (mean, 70 years). Representative morphologic and immunophenotypic features of the lymphomas are illustrated in supplemental Figure 1.
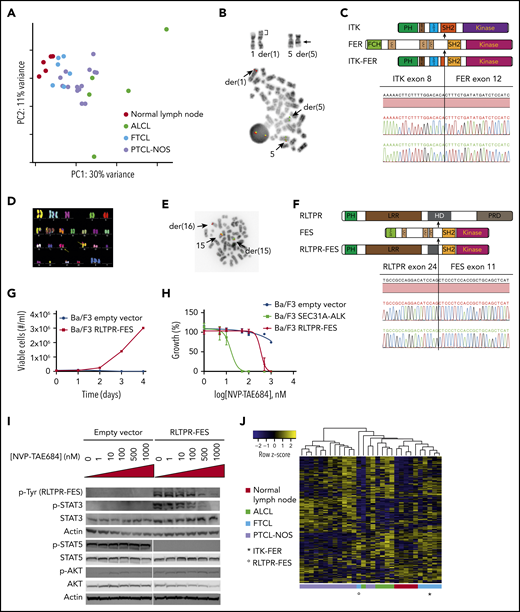
Genetic and functional analysis of the identified fusion genes. (A) Principal component (PC) analysis of gene counts for normal lymph nodes (n = 5), anaplastic large cell lymphoma (n = 5), FTCL (n = 6), and peripheral T-cell lymphoma not otherwise specified (n = 14) using our data and publically available data.23,24 (B) Partial karyotype illustrating inv(5)(q21q33) masked by t(1;5)(p34;q21) (breakpoints indicated by arrows) identified in patient 1 (upper panel) and metaphase FISH with a dual-color break-apart probe for FER (lower panel). (C) Schematic depiction and DNA sequence trace of the ITK-FER fusion. (D) Complex karyotype, including cryptic t(15;16)(q26;q22), identified by multicolor FISH in patient 2. (E) Metaphase FISH with a dual-color break-apart probe for FES performed in patient 2. (F) Schematic depiction and DNA sequence trace of the RLTPR-FES fusion. (G) Growth curve for Ba/F3 cells transduced with either empty vector or RLTPR-FES. (H) Relative proliferation of Ba/F3 cells transduced with empty vector, SEC31A-ALK, and RLTPR-FES in the presence of increasing concentrations of NVP-TAE684. (I) Western blot assessment of phosphorylated tyrosine residues (band at 131 kDa, corresponding to RLPTR-FES), STAT3 phosphorylation, STAT5 phosphorylation, and AKT phosphorylation in Ba/F3 cells transduced with empty vector or RLTPR-FES exposed to increasing concentrations of NVP-TA684. (J) Heat map representing the expression of STAT3 target genes in CD4+ T cells.
Genetic and functional analysis of the identified fusion genes. (A) Principal component (PC) analysis of gene counts for normal lymph nodes (n = 5), anaplastic large cell lymphoma (n = 5), FTCL (n = 6), and peripheral T-cell lymphoma not otherwise specified (n = 14) using our data and publically available data.23,24 (B) Partial karyotype illustrating inv(5)(q21q33) masked by t(1;5)(p34;q21) (breakpoints indicated by arrows) identified in patient 1 (upper panel) and metaphase FISH with a dual-color break-apart probe for FER (lower panel). (C) Schematic depiction and DNA sequence trace of the ITK-FER fusion. (D) Complex karyotype, including cryptic t(15;16)(q26;q22), identified by multicolor FISH in patient 2. (E) Metaphase FISH with a dual-color break-apart probe for FES performed in patient 2. (F) Schematic depiction and DNA sequence trace of the RLTPR-FES fusion. (G) Growth curve for Ba/F3 cells transduced with either empty vector or RLTPR-FES. (H) Relative proliferation of Ba/F3 cells transduced with empty vector, SEC31A-ALK, and RLTPR-FES in the presence of increasing concentrations of NVP-TAE684. (I) Western blot assessment of phosphorylated tyrosine residues (band at 131 kDa, corresponding to RLPTR-FES), STAT3 phosphorylation, STAT5 phosphorylation, and AKT phosphorylation in Ba/F3 cells transduced with empty vector or RLTPR-FES exposed to increasing concentrations of NVP-TA684. (J) Heat map representing the expression of STAT3 target genes in CD4+ T cells.
Clinical, pathological, and genetic features of the patients reported in this study
| Patient | Age, y | Immune profile | Sample at diagnosis | Karyotype* | FISH pattern | Mutation status | Stage† | Treatment/ response | Status | Survival, m |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 70 | CD4+, CD5+, CD7+, CD10+, BCL6+, PD1 (ICOS and CXCL13: NA), EBER+ | Lymph node | 46,XX,der(1)(5q35.3→5q33.3::5q21.3→5q33.3::1p34→1q44),der(5)t(1;5)(p34;q21.3) | SYK BA: 2F | NF | II | 2007: rituximab; CR; 2010/Rl: rituximab and mini-CHOP; NR; RT/PR; 2011/progression chlorambucil | Dead | 60 |
| FER BA: 1F1R1G | ||||||||||
| ITK BA: NI | ||||||||||
| 2 | 61 | CD4+, CD5+, CD7+, CD10+, BCL6+, PD1+, ICOS+, CXCL13+, EBER+ | Lymph node | 47,XX,+X,+2,del(6)(q11.1q22.3),+7,der(8)t(8;18)(p11.2;q12.1),der(15) t(15;16)(q26.1;q22.1),der(16)(20q13.3→20p13::16p13.3→p13.1::16p11.1→p13.1::16p13.1→q22.1::15q26.1→q26.3),–18,–20 | SYK BA: 2F | TET2H1380Y | II | CHOP, remains PET positive | Dead | 8 |
| ITK BA: 2F | TET2S1449fs | |||||||||
| FER BA: 2F | ||||||||||
| FES BA: 1F1R1G | ||||||||||
| 3 | 83 | CD4+, CD2–, CD7–, CD10+, BCL6–, PD1+, ICOS+, CXCL13+, EBER+ | Spleen | 46,X,–X,der(2)dup(2)(q33.1q33.3)t(2;11)(q37.3;q23.3),t(3;14)(p13;q32.2),del(5)(q14.2q23.2),del(6)(p21.31p24.1),der(8)t(8;8)(p11.21;q22.3),der(11)t(X;11)(q21.1;p15.5),+19 | SYK BA: 2F | RHOAG17V | IVBS | Splenectomy plus chlorambucil; progressive disease | Dead | 1 |
| ITK BA: 2F | IDH2R172K | |||||||||
| FER BA: 1F | TET2T1499fs | |||||||||
| FES BA: 2F | TET2Q1687X | |||||||||
| LSI IGH: 14F, der(3)F | ||||||||||
| 4 | 71 | CD3+, CD4+, CD5+, CD23+/ FDC, PD1+, CXCL13+, ICOS+, CD10p+, BCL6w+, EBER+ | Lymph node | 46,XX | SYK BA: 2F | RHOAG17V | III | CHOP; CR | Alive | 160 |
| ITK BA: 2F | IDH2R172S | |||||||||
| FER BA: 2F | TET2Q769X | |||||||||
| FES BA: 2F | TET2W1219fs | |||||||||
| DNMT3AR771P | ||||||||||
| 5 | 58 | CD3+, CD4+, CD5+, CD7–, CD10–, PD1+, ICOS+, BCL6+, CXCL13+, CD21+/FDC, EBER+ | Lymph node | 46,XX | SYK BA: 2F | RHOAG17V | IIIS | CHOP; CR | Alive | 111 |
| ITK BA: 2F | IDH2R172S | |||||||||
| FER BA: 2F | TET2N1714fs | |||||||||
| FES BA: 2F | TET2K1752fs | |||||||||
| 6 | 76 | CD3+, CD4+, PD1+, ICOS+, CXCL13+, CD10+, BCL6+, CD23+/FDC, EBER+ | Lymph node | 47,XX,+5 | SYK BA: 2F | RHOAG17V | IIIS | CHOP; CR | Alive | 20 |
| ITK BA: 3F | IDH2R172M | |||||||||
| FER BA: 3F | TET2Y1294C | |||||||||
| FES BA: 2F |
| Patient | Age, y | Immune profile | Sample at diagnosis | Karyotype* | FISH pattern | Mutation status | Stage† | Treatment/ response | Status | Survival, m |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 70 | CD4+, CD5+, CD7+, CD10+, BCL6+, PD1 (ICOS and CXCL13: NA), EBER+ | Lymph node | 46,XX,der(1)(5q35.3→5q33.3::5q21.3→5q33.3::1p34→1q44),der(5)t(1;5)(p34;q21.3) | SYK BA: 2F | NF | II | 2007: rituximab; CR; 2010/Rl: rituximab and mini-CHOP; NR; RT/PR; 2011/progression chlorambucil | Dead | 60 |
| FER BA: 1F1R1G | ||||||||||
| ITK BA: NI | ||||||||||
| 2 | 61 | CD4+, CD5+, CD7+, CD10+, BCL6+, PD1+, ICOS+, CXCL13+, EBER+ | Lymph node | 47,XX,+X,+2,del(6)(q11.1q22.3),+7,der(8)t(8;18)(p11.2;q12.1),der(15) t(15;16)(q26.1;q22.1),der(16)(20q13.3→20p13::16p13.3→p13.1::16p11.1→p13.1::16p13.1→q22.1::15q26.1→q26.3),–18,–20 | SYK BA: 2F | TET2H1380Y | II | CHOP, remains PET positive | Dead | 8 |
| ITK BA: 2F | TET2S1449fs | |||||||||
| FER BA: 2F | ||||||||||
| FES BA: 1F1R1G | ||||||||||
| 3 | 83 | CD4+, CD2–, CD7–, CD10+, BCL6–, PD1+, ICOS+, CXCL13+, EBER+ | Spleen | 46,X,–X,der(2)dup(2)(q33.1q33.3)t(2;11)(q37.3;q23.3),t(3;14)(p13;q32.2),del(5)(q14.2q23.2),del(6)(p21.31p24.1),der(8)t(8;8)(p11.21;q22.3),der(11)t(X;11)(q21.1;p15.5),+19 | SYK BA: 2F | RHOAG17V | IVBS | Splenectomy plus chlorambucil; progressive disease | Dead | 1 |
| ITK BA: 2F | IDH2R172K | |||||||||
| FER BA: 1F | TET2T1499fs | |||||||||
| FES BA: 2F | TET2Q1687X | |||||||||
| LSI IGH: 14F, der(3)F | ||||||||||
| 4 | 71 | CD3+, CD4+, CD5+, CD23+/ FDC, PD1+, CXCL13+, ICOS+, CD10p+, BCL6w+, EBER+ | Lymph node | 46,XX | SYK BA: 2F | RHOAG17V | III | CHOP; CR | Alive | 160 |
| ITK BA: 2F | IDH2R172S | |||||||||
| FER BA: 2F | TET2Q769X | |||||||||
| FES BA: 2F | TET2W1219fs | |||||||||
| DNMT3AR771P | ||||||||||
| 5 | 58 | CD3+, CD4+, CD5+, CD7–, CD10–, PD1+, ICOS+, BCL6+, CXCL13+, CD21+/FDC, EBER+ | Lymph node | 46,XX | SYK BA: 2F | RHOAG17V | IIIS | CHOP; CR | Alive | 111 |
| ITK BA: 2F | IDH2R172S | |||||||||
| FER BA: 2F | TET2N1714fs | |||||||||
| FES BA: 2F | TET2K1752fs | |||||||||
| 6 | 76 | CD3+, CD4+, PD1+, ICOS+, CXCL13+, CD10+, BCL6+, CD23+/FDC, EBER+ | Lymph node | 47,XX,+5 | SYK BA: 2F | RHOAG17V | IIIS | CHOP; CR | Alive | 20 |
| ITK BA: 3F | IDH2R172M | |||||||||
| FER BA: 3F | TET2Y1294C | |||||||||
| FES BA: 2F |
BA, break-apart; CHOP, cyclophosphamide, doxorubicin, vincristine, and prednisone; CR, complete remission; F, fused signal; FDC, follicular dendritic cell; G, green signal; NA, not available; NF, not found; NI, not informative because of a poor resolution of signals on inv(5); NR, no response; p, partial; PET, positron emission tomography; PR, partial response; R, red signal; Rl, relapse; RT, radiotherapy; w, weak.
Corrected after FISH and array comparative genomic hybridization studies (data not shown).
Staging according to Ann Arbor.
The karyotype of patient 1 showed a sole t(1;5)(p34;q21.3). FISH identified involvement of the FER gene (5q21.3) (Figure 1B) and revealed that t(1;5)(p34;q21.3) in fact masked a cryptic inv(5)(q21.3q33.3). RNA-seq identified an in-frame fusion of exon 8 of ITK (5q33.3) to exon 12 of FER. The fusion was confirmed by reverse transcription polymerase chain reaction and Sanger sequencing (Figure 1C). Patient 2 harbored complex numerical and structural rearrangements, as demonstrated by multicolor FISH analysis (Figure 1D). FISH analysis with break-apart probes for 14 candidate protein tyrosine kinase (PTK) genes (supplemental Table 1) neighboring chromosomal rearrangements identified a breakpoint in FES at 15q26.1 (Figure 1E). RNA-seq identified an in-frame fusion of exon 24 of RLTPR (RGD, leucine-rich repeat, tropomodulin and proline-rich containing protein) at 16q22.1 to exon 11 of FES. The t(15;16)(q26.1;q22.1)/RLTPR-FES rearrangement was confirmed by reverse transcription polymerase chain reaction and Sanger sequencing (Figure 1F). Molecular cytogenetics and RNA-seq of the remaining 4 patients have not identified any PTK fusion genes. By using RNA-seq data, we analyzed the mutation status of genes that are recurrently mutated in TFH-derived lymphomas (TET2, DNMT3A, RHOA, IDH2, CD28, FYN, and VAV14,7 ). None of them were mutated in patient 1 with ITK-FER, whereas patient 2 with RLTPR-FES harbored TET2 mutations. The 4 PTK fusion-negative patients carried the RHOAG17V and IDH2R172 mutations as well as TET2 mutations (Table 1).
Previous studies showed that ITK-SYK and ITK-FER fusions act as constitutive tyrosine kinases. ITK-SYK mimics T-cell receptor (TCR) signaling6 and both ITK-SYK and ITK-FER phosphorylate STAT3.8 To determine the oncogenic properties of the novel RLTPR-FES fusion protein, we expressed RLTPR-FES in the murine hematopoietic IL-3–dependent Ba/F3 cell line. Upon IL-3 withdrawal, RLTPR-FES conferred growth factor–independent growth, revealing that this fusion protein is constitutively active and supports the proliferation and survival of Ba/F3 cells (Figure 1G). Next, we investigated the ability of NVP-TAE684 (small molecule ATP-competitive ALK/FES inhibitor9,10 ) to inhibit the activity of the RLTPR-FES kinase. Ba/F3 cells transformed by RLTPR-FES responded in a dose-dependent manner to NVP-TAE684 treatment and were slightly less sensitive to the inhibitor than SEC31A-ALK–expressing Ba/F3 cells11 (Figure 1H). A direct inhibitory effect of NVP-TAE684 on RLTPR-FES kinase activity was confirmed by western blot analysis using phospho-specific antibodies. Furthermore, phosphorylation of STAT3 was reduced upon treatment with NVP-TAE684. There was no effect on phosphorylation of AKT, ERK1/2, and STAT5 (Figure 1I). Immunohistochemistry confirmed high levels of STAT3 phosphorylation in the presence of RLTPR-FES and ITK-FER in the biopsies (supplemental Figure 2). The heterogeneous tumor composition (supplemental Figure 3A), did not allow the identification of a STAT3 signature in bulk RNA (supplemental Figure 3B; supplemental Table 4). We used CIBERSORTx to impute the expression of STAT3 target genes in CD4+ T cells from bulk RNA (Figure 1J). We confirmed high expression levels of STAT3 target genes in patient 2 (RLTPR-FES). This was less evident in patient 1 (ITK-FER), but this patient had the lowest ratio of TFH cells over total CD4+ T cells.
FER and FES are the only members of a subfamily of nonreceptor PTKs. FER is targeted by at least 3 tumor-related fusions: SSBP2-FER identified in a patient with T-cell acute lymphoblastic leukemia,12 MAN2A1-FER found in hepatocellular carcinoma and other cancers,13 and ITK-FER detected in 1 patient with peripheral T-cell lymphoma not otherwise specified.14 All fusions resulted in aberrant constitutive tyrosine kinase activity of FER, and their oncogenic potential was demonstrated in vitro. In contrast, FES seems to play a dual role in tumorigenesis, acting as an oncogene or a tumor suppressor, depending on the cellular context.15,16 RLTPR, partner of FES, codes for proteins involved in cytoskeletal organization and cell migration and is required for CD28 costimulation in T cells.17 The RLTPR-FES fusion contains the RLTPR homodimerization domain,18 which is presumably important for the enhanced kinase activity of chimera, because FES oligomerization enhances FES kinase activity.19
The occurrence of multiple mutations in patients with FTCL is in line with the proposed model of multistage development of TFH-derived lymphomas.7 These tumors seem to be driven by 2 types of cooperating mutations: (1) early premalignant (nonlineage impact) mutations affecting genes involved in the regulation of DNA methylation (TET2, DNMT3A, IDH2), which occur in hematopoietic stem cells and confer proliferative advantage, and (2) tumor-specific (lineage impact) mutations targeting genes critical to T-cell biology. TFH differentiation requires sustained TCR signaling, costimulation through ICOS and the IL-21-STAT3 axis.20 RHOA mutations enhance ICOS signaling,21 and ITK-SYK mimics a TCR signal.6 RLTPR-FES and ITK-FER represent genetic hits that hijack STAT3 signaling to drive TFH-derived lymphoma.
Our study supports the critical role of PTK fusion genes in the pathogenesis of FTCL and provides additional evidence that these can drive FTCL in the absence of RHOA mutations. Importantly, we showed that TFH-derived lymphoma can be driven by oncogenic activation of the STAT3 axis. Given that PTKs are amenable for targeted therapy and that murine RHOA-driven lymphomas are responsive to the PI3K inhibitor duvelisib21 and the mTOR inhibitor everolimus,22 the use of new therapeutic agents in FTCL should be considered.
RNA-seq data are available at Gene Expression Omnibus (Accession No. GSE57944 for patients 1 to 3; pending for patients 4 to 6).
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Kathleen Doms, Ursula Pluys, Kim Rummens, and Vanessa Vanspauwen for their excellent technical assistance and Rita Logist for editorial help.
This study was supported by the concerted action grant from the Katholieke Universiteit Leuven No. 3M040406 (J.-A.v.d.K., P.V., T.T., J.C., and I.W.). K.D. was supported by an aspirant fellowship of Fonds voor Wetenschappelijk Onderzoek-Vlaanderen. T.T. holds a Mandate for Fundamental and Translational Research from the “Stichting tegen Kanker” (2°14-083). D.D. holds a Mandate for Clinical and Translational Research from “Kom op tegen Kanker” and for Clinical Research from the University Hospitals Leuven. P.V. is a senior clinical investigator of the Fonds voor Wetenschappelijk Onderzoek-Vlaanderen.
Authorship
Contribution: K.D., J.-A.v.d.K., T.T., J.C., and I.W. designed and performed the research, analyzed the data, and wrote the manuscript; J.A.F.F., K.V.R., L. Marcelis, and G.A. performed research and analyzed data; C.G., D.D., P.V., and L. Michaux provided and analyzed clinical data; and all authors reviewed and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Iwona Wlodarska, Center for Human Genetics, Herestr 49, B-3000 Leuven, Belgium; e-mail: iwona.wlodarska@uzleuven.be.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal