

Key Points
DARA SC-CyBorD was well tolerated in patients (excluded Mayo stage IIIb) with newly diagnosed AL amyloidosis.
DARA SC-CyBorD elicited robust hematologic and organ responses in these patients.

Abstract
Although no therapies are approved for light chain (AL) amyloidosis, cyclophosphamide, bortezomib, and dexamethasone (CyBorD) is considered standard of care. Based on outcomes of daratumumab in multiple myeloma (MM), the phase 3 ANDROMEDA study (NCT03201965) is evaluating daratumumab-CyBorD vs CyBorD in newly diagnosed AL amyloidosis. We report results of the 28-patient safety run-in. Patients received subcutaneous daratumumab (DARA SC) weekly in cycles 1 to 2, every 2 weeks in cycles 3 to 6, and every 4 weeks thereafter for up to 2 years. CyBorD was given weekly for 6 cycles. Patients had a median of 2 involved organs (kidney, 68%; cardiac, 61%). Patients received a median of 16 (range, 1-23) treatment cycles. Treatment-emergent adverse events were consistent with DARA SC in MM and CyBorD. Infusion-related reactions occurred in 1 patient (grade 1). No grade 5 treatment-emergent adverse events occurred; 5 patients died, including 3 after transplant. Overall hematologic response rate was 96%, with a complete hematologic response in 15 (54%) patients; at least partial response occurred in 20, 22, and 17 patients at 1, 3, and 6 months, respectively. Renal response occurred in 6 of 16, 7 of 15, and 10 of 15 patients, and cardiac response occurred in 6 of 16, 6 of 13, and 8 of 13 patients at 3, 6, and 12 months, respectively. Hepatic response occurred in 2 of 3 patients at 12 months. Daratumumab-CyBorD was well tolerated, with no new safety concerns versus the intravenous formulation, and demonstrated robust hematologic and organ responses. This trial was registered at www.clinicaltrials.gov as #NCT03201965.
Introduction
Systemic amyloid light-chain (AL) amyloidosis is a rare plasma cell disorder primarily affecting older adults. In the United States, the unadjusted incidence is approximately 10 to 14 cases per million person-years,1 which is likely underestimated because of delayed or missed diagnosis. AL amyloidosis is characterized by deposition of insoluble amyloid fibrils into tissues and organs, resulting in progressive organ damage. Affected organs most frequently include the heart, kidney, and liver, but soft tissues and the nervous system may be involved.2,3
Application of novel drugs developed for multiple myeloma (MM), and in particular bortezomib, have improved AL amyloidosis outcomes.4,5 Among patients at the Mayo Clinic in the United States, the 2-year overall survival (OS) rate increased from 42% among those diagnosed from 2000 to 2004 to 60% in patients diagnosed from 2010 to 2014.5 In a population-based Swedish study, the 2-year OS rate improved from 42% to 61% between 2000 to 2004 and 2010 to 2013.6 Outcomes in both studies suggested that early diagnosis and treatment with more effective antiplasma cell therapy could decrease early mortality.
Despite these promising findings, antiplasma cell therapy remains suboptimal for most patients with AL amyloidosis. Hematologic complete response (CR) rates in newly diagnosed patients receiving commonly used drug regimens such as cyclophosphamide, bortezomib, and dexamethasone (CyBorD) range from 23% to 47%.7,8 Similar or higher CR rates are achievable with high-dose melphalan treatment and autologous stem cell transplant (ASCT), but this therapy is only feasible in a minority of patients.9-11 In addition, patients with AL amyloidosis experience more frequent and severe toxicity compared with patients with MM receiving the same regimens.12,13 Thus, an unmet need remains for more tolerable and effective therapies for AL amyloidosis.
Depth of hematologic response is strongly associated with organ response and improved survival in AL amyloidosis.14 Antiplasma cell regimens that induce rapid, deep, and durable hematologic responses can ameliorate organ dysfunction and ultimately increase OS. Daratumumab is a human immunoglobulin Gκ (IgGκ) monoclonal antibody targeting CD38 that is uniformly expressed on clonal plasma cells and has a direct on-tumor and immunomodulatory mechanism of action.15-21 In MM, daratumumab (16 mg/kg intravenous [IV]) has demonstrated efficacy as monotherapy and in combination with standard regimens in newly diagnosed and relapsed MM.22-28 Daratumumab combination regimens have shown remarkable rates of undetectable minimal residual disease and a predictable and manageable safety profile,26-30 and have not been associated with cardiac or renal toxicities, which are of particular concern to patients with AL amyloidosis.22-26
AL amyloidosis plasma cells have been shown to express CD38.31,32 In addition, preliminary results of 2 prospective studies of daratumumab monotherapy in relapsed AL amyloidosis have shown promising hematologic responses without cardiac, renal, or other notable toxicities and overall hematologic response rates of at least 59%.33-35 These promising results and favorable attributes make daratumumab ideally suited for study in the AL population with compromised organ function.
Here we present for the first time the use of the subcutaneous formulation of daratumumab (DARA SC) in AL amyloidosis in the safety run-in cohort of the phase 3 ANDROMEDA study (AMY3001; ClinicalTrials.gov identifier: NCT03201965). This study is investigating DARA SC in combination with CyBorD in patients with newly diagnosed AL amyloidosis.
Methods
Study design
ANDROMEDA is a randomized, open-label, active-controlled, multicenter, phase 3 study with a safety run-in phase. Here we report the results of the safety run-in phase, which was conducted to determine the safety and tolerability of DARA SC plus CyBorD in at least 10 patients with newly diagnosed AL amyloidosis. If no safety signals were observed after at least 1 cycle of treatment, the randomized portion of the study would begin with approximately 360 patients being randomly assigned 1:1 to receive CyBorD with or without DARA SC. All patients in the safety run-in cohort received DARA SC (1800 mg in 15 mL) with recombinant human hyaluronidase PH20 (rHuPH20; 30 000 U; ENHANZE drug delivery technology; Halozyme, Inc).
DARA SC was administered in a single, premixed vial and given by manual SC injection over the course of 3 to 5 minutes weekly in cycles 1 to 2, every 2 weeks in cycles 3 to 6, and every 4 weeks thereafter as monotherapy for a maximum of 2 years from study start (all cycles were 28 days). Cyclophosphamide 300 mg/m2 orally or intravenously and bortezomib 1.3 mg/m2 subcutaneously were given on days 1, 8, 15, and 22 of each cycle for up to 6 cycles. Dexamethasone 40 mg (starting dose) was given orally or intravenously weekly for each cycle for up to 6 cycles. For patients who were older than 70 years, underweight (body mass index <18.5 kg/m2), or had hypervolemia (including heart failure), poorly controlled diabetes mellitus, or prior intolerance to steroid therapy, dexamethasone could be administered at 20 mg weekly per investigator discretion.
Patients
Eligible patients were at least 18 years of age with a histopathologic diagnosis of systemic AL amyloidosis and measurable hematologic disease without prior therapy. Histopathologic diagnosis of amyloidosis was based on detection by immunohistochemistry and polarizing light microscopy of green birefringent material in tissue specimens stained with Congo red in an organ other than bone marrow, or characteristic electron microscopy appearance (unbranched 10-nm-thick fibrils). Individuals whose only evidence of amyloid deposition was in the bone marrow were excluded. Age-related amyloidosis and hereditary amyloidosis were ruled out, respectively, in male patients at least 70 years of age with cardiac involvement only, and in patients of African descent, using mass spectrometry typing of amyloid deposits in a tissue biopsy.
Measurable disease was defined by either a serum free light-chain (FLC) level of at least 50 mg/L with an abnormal κ to λ ratio or a difference between involved (amyloidogenic) FLC (iFLC) and uninvolved FLC (uFLC) of at least 50 mg/L; or a serum monoclonal protein level of at least 5.0 g/L. During the study screening phase, patients must have had an absolute neutrophil count of at least 1.0 × 109/L, hemoglobin levels of at least 80 g/L, platelet count of at least 50 × 109/L, aspartate and alanine aminotransferase levels ≤2.5 times the upper limit of normal (ULN), and total bilirubin levels ≤1.5 times the ULN. Estimated glomerular filtration rate, as determined by the Chronic Kidney Disease Epidemiology Collaboration equation, was required to be at least 20 mL/min/1.73 m2. Eligible patients had at least 1 affected organ according to consensus criteria for the organ involved,36,37 and an Eastern Cooperative Oncology Group38 performance status score of ≤2. Eligible patients were classified by cardiac stage at screening based on the European Modification of the Mayo Clinical Cardiac Staging system.39 This system categorizes patients by the presence of 2 risk factors defined by elevated levels of the biomarkers N-terminal probrain natriuretic peptide (NT-proBNP: >332 ng/L) and high-sensitivity cardiac troponin (>54 ng/L).40 Stage I patients had neither risk factor, stage II patients had 1 risk factor, stage IIIa patients had both risk factors with NT-proBNP levels 8500 ng/L or less, and stage IIIb patients had NT-proBNP levels higher than 8500 ng/L.41
Patients were not eligible if they had prior therapy for AL amyloidosis or MM (including CD38-targeted agents) or had a previous or current diagnosis of symptomatic MM. Patients with significant cardiovascular conditions, as evidenced by NT-proBNP levels higher than 8500 ng/L, New York Heart Association classification IIIb or IV heart failure,42 ischemic heart disease or uncorrected valvular disease unrelated to AL amyloid cardiomyopathy, sustained ventricular tachycardia, aborted ventricular fibrillation, atrioventricular nodal or sinoatrial nodal dysfunction with no pacemaker, QT interval as corrected by Fridericia’s formula higher than 500 msec without pacemaker, supine systolic blood pressure lower than 90 mm Hg, or symptomatic orthostatic hypotension (a decrease in systolic blood pressure upon standing of >20 mm Hg despite medical management [eg, midodrine, fludrocortisones] in the absence of volume depletion) were excluded. Patients with a history of malignancy (other than AL amyloidosis), chronic obstructive pulmonary disease, moderate or severe persistent asthma, current uncontrolled asthma, positivity for human immunodeficiency virus, active hepatitis B or C infection, grade 2 sensory or grade 1 painful peripheral neuropathy, any form of non-AL amyloidosis, or any concurrent medical condition or disease that would likely interfere with study procedures or results were excluded.
Study endpoints
In the safety run-in phase, absence of a safety signal (particularly with regard to volume overload) was required for the randomized portion of the study to begin. The primary endpoint of the randomized portion of ANDROMEDA is overall complete hematologic response rate (CR) based on International Amyloidosis Consensus Criteria guidelines.14,43,44 Amyloidosis CR (aCR) criteria require normalization of FLC levels and ratio, and negative serum and urine immunofixation (IFE). Patients who demonstrated negative serum and urine IFE but did not achieve a normalized FLC ratio because of the suppression of uFLC below the lower limit of normal (FLC ratio abnormal or normal) and who achieved normalized iFLC levels could not be formally categorized as achieving aCR and were classified as having a modified CR (mCR). Among patients with measurable difference between involved and uninvolved FLC (dFLC ≥50 mg/L), very good partial response (VGPR) was defined as a reduction of dFLC to less than 40 mg/L, and a PR by a decrease in dFLC by more than 50%.
A secondary endpoint was major organ deterioration progression-free survival, a composite of endpoints occurring from randomization to whichever of the following occurs first: death, clinical manifestation of cardiac or renal failure, or hematologic progressive disease per consensus guidelines. Cardiac failure was defined as development of dyspnea at rest for at least 3 consecutive days solely as a result of amyloidosis cardiac deterioration or need for left ventricular assist device, intra-aortic balloon pump, or cardiac transplant. Renal failure was defined as development of end-stage renal disease requiring hemodialysis or renal transplant. Hematologic progression was defined on the basis of International Amyloidosis Consensus Criteria: starting from CR, a change to abnormal FLC ratio (iFLC must double and be above ULN) or reappearance of the involved monoclonal protein on IFE; starting from CR/VGPR/PR, a 50% increase in serum M-protein to more than 0.5 g/dL or a 50% increase in urine M-protein to more than 200 mg/day (visible peak must be present); or FLC increase of 50% to more than 100 mg/L.
Other secondary endpoints included organ response rate (assessed by biomarkers), progression-free survival, OS, improvement in patient-reported fatigue according to the European Organization for Research and Treatment of Cancer Quality of Life Questionnaire, time to next treatment, rate of hematologic VGPR or better, time to hematologic and organ response, and duration of organ response. Cardiac response was defined as more than 30% and more than 300 ng/L decreases in NT-proBNP levels in patients with baseline levels of at least 650 ng/L.14 Cardiac progression was defined as more than 30% and more than 300 ng/L increases in NT-proBNP levels, at least 33% increase in cardiac troponin levels, or at least 10% decrease in left ventricular ejection fraction. Renal response was characterized by at least a 30% decrease in proteinuria or decrease in proteinuria below 0.5 g in 24 hours without renal progression. Renal progression was defined as at least a 25% decrease in estimated glomerular filtration rate. Hepatic responses were defined as a 50% decrease in abnormal alkaline phosphatase values; progression of liver disease was considered a 50% increase in alkaline phosphatase level above the lowest value.14
Study analyses
In the safety run-in, safety was evaluated after at least 10 patients received 1 or more treatment cycles. Treatment-emergent adverse events (TEAEs) were coded using the Medical Dictionary for Regulatory Activities. Dosing was staggered 48 hours or more between patients to assess infusion-related reactions (IRRs). Preliminary overall best hematologic response rates were also evaluated. All continuous variables were summarized using descriptive statistics, whereas categorical variables were summarized using frequency and percentage, unless stated otherwise.
Hematologic responses were evaluated weekly for cycle 1, every 4 weeks for cycles 2 to 6, and every other month thereafter until major organ deterioration progression-free survival, death, withdrawal of consent to participate, or end of the study. Organ responses were categorized by increased functionality measured by organ-specific serum and urine assays,14,45 and were assessed according to the same schedule as hematologic responses. Cardiac response was evaluated at a central laboratory.
Study oversight
The study was registered at ClinicalTrials.gov (NCT03201965), and was sponsored by Janssen Research & Development, LLC. Institutional review boards or independent ethics committees at study sites approved this study. Each patient provided written consent according to local requirements. The investigators and sponsor devised the study design and analysis. Study data were collected by investigators and their research teams. Janssen conducted the final data analysis and verified data accuracy. Investigators were not restricted by confidentiality agreements and had full accessibility to all data.
Results
Patients and treatment
A total of 28 patients received at least 1 treatment cycle in the safety run-in portion of the study. Patient demographics and baseline characteristics are presented in Table 1. Median age was 67.5 (range, 35-83) years, with more than half (n = 16 [57%]) of patients aged at least 65 years. Median time from diagnosis was 59.5 (range, 15-501) days. Twenty-two (79%) patients had measurable disease, as indicated by serum FLC levels only, and 3 (11%) patients each had disease as measured by serum M-protein levels only and by both serum M-protein and FLC levels. FLC isotypes were λ (75%) and κ (25%). Immunoglobulin heavy chain expression was observed in 10 patients (8 [30%] IgG and 2 [7%] IgA). More than 50% of patients had at least 2 organs involved, with 61% and 68% of the patients having heart and kidney involvement, respectively. The majority (n = 22 [79%]) of patients were classified as cardiac stage II or higher per the modified Mayo staging system.39 One patient with values corresponding to stage IIIa during screening subsequently increased to IIIb on cycle 1 day 1.
Demographics and baseline characteristics
| Characteristic | Patients (n = 28) |
|---|---|
| Age | |
| Median (range), years | 67.5 (35-83) |
| Category, n (%) | |
| <65 years | 12 (42.9) |
| ≥65 years | 16 (57.1) |
| Male, n (%) | 16 (57.1) |
| Race, n (%) | |
| White | 25 (89.3) |
| Black/African American | 2 (7.1) |
| Unknown | 1 (3.6) |
| ECOG performance status,*n (%) | |
| 0 | 7 (25.0) |
| 1 | 18 (64.3) |
| 2 | 3 (10.7) |
| Time from diagnosis | |
| Median (range), days | 59.5 (15-501) |
| Involved organs, n (%) | |
| Median, n (range) | 2 (1-4) |
| ≥2 organs | 19 (67.9) |
| Kidney | 19 (67.9) |
| Heart | 17 (60.7) |
| Nerve | 6 (21.4) |
| Gastrointestinal tract | 5 (17.9) |
| Peripheral nervous system | 5 (17.9) |
| Liver | 4 (14.3) |
| Soft tissue | 4 (14.3) |
| Autonomic nervous system | 1 (3.6) |
| FLC isotype, n (%) | |
| Λ | 21 (75.0) |
| κ | 7 (25.0) |
| Immunoglobin heavy chain isotype, n (%) | N = 27 |
| Any heavy chain expression | 10 (37.0) |
| IgG | 8 (29.6) |
| IgA | 2 (7.4) |
| Mayo Clinic cardiac stage,†n (%) | |
| I | 6 (21.4) |
| II | 16 (57.1) |
| IIIa | 5 (17.9) |
| IIIb‡ | 1 (3.6) |
| NYHA class,§n (%) | |
| I | 17 (60.7) |
| II | 10 (35.7) |
| IIIA | 1 (3.6) |
| Baseline creatinine clearance, n (%) | |
| n | 27 |
| ≥60 mL/minute | 20 (74.1) |
| <60 mL/minute | 7 (25.9) |
| Characteristic | Patients (n = 28) |
|---|---|
| Age | |
| Median (range), years | 67.5 (35-83) |
| Category, n (%) | |
| <65 years | 12 (42.9) |
| ≥65 years | 16 (57.1) |
| Male, n (%) | 16 (57.1) |
| Race, n (%) | |
| White | 25 (89.3) |
| Black/African American | 2 (7.1) |
| Unknown | 1 (3.6) |
| ECOG performance status,*n (%) | |
| 0 | 7 (25.0) |
| 1 | 18 (64.3) |
| 2 | 3 (10.7) |
| Time from diagnosis | |
| Median (range), days | 59.5 (15-501) |
| Involved organs, n (%) | |
| Median, n (range) | 2 (1-4) |
| ≥2 organs | 19 (67.9) |
| Kidney | 19 (67.9) |
| Heart | 17 (60.7) |
| Nerve | 6 (21.4) |
| Gastrointestinal tract | 5 (17.9) |
| Peripheral nervous system | 5 (17.9) |
| Liver | 4 (14.3) |
| Soft tissue | 4 (14.3) |
| Autonomic nervous system | 1 (3.6) |
| FLC isotype, n (%) | |
| Λ | 21 (75.0) |
| κ | 7 (25.0) |
| Immunoglobin heavy chain isotype, n (%) | N = 27 |
| Any heavy chain expression | 10 (37.0) |
| IgG | 8 (29.6) |
| IgA | 2 (7.4) |
| Mayo Clinic cardiac stage,†n (%) | |
| I | 6 (21.4) |
| II | 16 (57.1) |
| IIIa | 5 (17.9) |
| IIIb‡ | 1 (3.6) |
| NYHA class,§n (%) | |
| I | 17 (60.7) |
| II | 10 (35.7) |
| IIIA | 1 (3.6) |
| Baseline creatinine clearance, n (%) | |
| n | 27 |
| ≥60 mL/minute | 20 (74.1) |
| <60 mL/minute | 7 (25.9) |
ECOG, Eastern Cooperative Oncology Group; NYHA, New York Heart Association.
ECOG performance status is scored on a scale from 0 to 5, with 0 indicating no symptoms and higher scores indicating increasing disability.
Based on the European Modification of the Mayo staging system39 ; cardiac stage was based on 2 biomarker risk factors: NT-proBNP and high sensitivity cardiac troponin.
One patient with values corresponding to IIIa during screening subsequently increased to IIIb on cycle 1 day 1.
NYHA classification class I patients have no limitation during ordinary physical activity, class II patients have slight limitation during ordinary physical activity, class IIIA patients have symptoms with less than ordinary physical activity, class IIIb patients have symptoms with daily living activities, and class IV patients have symptoms at rest.42 Patients with class IIb or IV disease were excluded from the study.
Patients received a median of 16 (range, 1-23) treatment cycles with a median duration of treatment of 15.1 (range, 0.2-20.1) months. Median dose intensities were 80% (range, 62%-99%) for cyclophosphamide, 96% (range 56%-101%) for bortezomib, 97% (range, 52%-102%) for dexamethasone, and 100% (range, 85%-100%) for DARA SC. The median duration of the first DARA SC injection was 5 (range, 3-17) minutes; second and subsequent injections also had a median duration of 5 minutes (respective ranges, 4-9 and 1-15 minutes). The median duration of follow-up was 17.6 (range, 1.3-20.4) months. A total of 25 (89%) patients have received at least 6 cycles of treatment; 3 patients received all 6 planned cycles of DARA SC plus CyBorD without subsequent DARA SC maintenance, and 22 (79%) patients received DARA SC maintenance monotherapy (>6 treatment cycles). At the time of clinical cutoff (23 July 2019), a total of 13 (46%) patients had discontinued treatment.
A total of 9 (32%) patients underwent elective ASCT (Figure 1). These patients received a median of 7 (range, 6-16) cycles of DARA SC plus CyBorD or DARA SC monotherapy. All patients were able to mobilize adequate numbers of CD34+ cells (median, 7 × 106/kg; range, 3-14 × 106/kg), all were mobilized with granulocyte colony-stimulating factor or equivalent, and 6 were also given plerixafor. One patient required a second mobilization attempt.
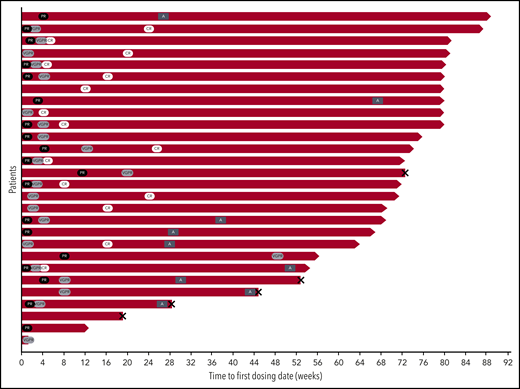
Swim lane plot of patients enrolled in the safety run-in portion of ANDROMEDA. Patient disposition by hematologic response is shown for the 28 patients enrolled in the safety run-in portion of the study. Black ovals indicate PR, light gray ovals indicate VGPR, and white ovals indicate CR. Dark gray rectangles indicate ASCT, and X indicates death. A, autologous stem cell transplant.
Swim lane plot of patients enrolled in the safety run-in portion of ANDROMEDA. Patient disposition by hematologic response is shown for the 28 patients enrolled in the safety run-in portion of the study. Black ovals indicate PR, light gray ovals indicate VGPR, and white ovals indicate CR. Dark gray rectangles indicate ASCT, and X indicates death. A, autologous stem cell transplant.
Safety
The most common any-grade and grade 3/4 TEAEs are reported in Table 2. Twenty-six (93%) patients experienced TEAEs considered related to study treatment; TEAEs in 21 (75%) patients were considered related to daratumumab. A total of 14% of patients experienced any-grade peripheral sensory neuropathy with DARA SC plus CyBorD (no grade 3/4 events). Grade 3/4 infections included pneumonia (n = 3 [11%]), cellulitis (n = 2 [7%]), and peritonitis, upper respiratory tract infection, and vascular device infection (n = 1 each [4%]).
Most common all-grade TEAEs and grade 3/4 TEAEs
| Patients (n = 28) | ||
|---|---|---|
| All-grade TEAEs (≥25%), n (%) | Grade 3/4 TEAEs (>1 patient), n (%) | |
| Overall | 28 (100) | 20 (71.4) |
| Diarrhea | 19 (67.9) | 4 (14.3) |
| Fatigue | 15 (53.6) | 6 (21.4) |
| Peripheral edema | 14 (50.0) | 4 (14.3) |
| Anemia | 13 (46.4) | 4 (14.3) |
| Constipation | 13 (46.4) | 0 |
| Dizziness | 13 (46.4) | 0 |
| Lymphopenia | 12 (42.9) | 5 (17.9) |
| Nausea | 12 (42.9) | 0 |
| Upper respiratory tract infection | 11 (39.3) | 1 (3.6) |
| Hyperglycemia | 10 (35.7) | 0 |
| Insomnia | 9 (32.1) | 1 (3.6) |
| Dyspnea | 9 (32.1) | 0 |
| Cough | 8 (28.6) | 0 |
| Hypoalbuminemia | 7 (25.0) | 2 (7.1) |
| Hyponatremia | 7 (25.0) | 2 (7.1) |
| Hypokalemia | 7 (25.0) | 1 (3.6) |
| Aspartate aminotransferase increased | 7 (25.0) | 0 |
| Thrombocytopenia | 7 (25.0) | 1 (3.6) |
| Fall | 6 (21.4) | 3 (10.7) |
| Cellulitis | 4 (14.3) | 2 (7.1) |
| Acute kidney injury | 3 (10.7) | 2 (7.1) |
| Pneumonia | 3 (10.7) | 3 (10.7) |
| Hypertension | 2 (7.1) | 1 (3.6) |
| Syncope | 2 (7.1) | 2 (7.1) |
| Patients (n = 28) | ||
|---|---|---|
| All-grade TEAEs (≥25%), n (%) | Grade 3/4 TEAEs (>1 patient), n (%) | |
| Overall | 28 (100) | 20 (71.4) |
| Diarrhea | 19 (67.9) | 4 (14.3) |
| Fatigue | 15 (53.6) | 6 (21.4) |
| Peripheral edema | 14 (50.0) | 4 (14.3) |
| Anemia | 13 (46.4) | 4 (14.3) |
| Constipation | 13 (46.4) | 0 |
| Dizziness | 13 (46.4) | 0 |
| Lymphopenia | 12 (42.9) | 5 (17.9) |
| Nausea | 12 (42.9) | 0 |
| Upper respiratory tract infection | 11 (39.3) | 1 (3.6) |
| Hyperglycemia | 10 (35.7) | 0 |
| Insomnia | 9 (32.1) | 1 (3.6) |
| Dyspnea | 9 (32.1) | 0 |
| Cough | 8 (28.6) | 0 |
| Hypoalbuminemia | 7 (25.0) | 2 (7.1) |
| Hyponatremia | 7 (25.0) | 2 (7.1) |
| Hypokalemia | 7 (25.0) | 1 (3.6) |
| Aspartate aminotransferase increased | 7 (25.0) | 0 |
| Thrombocytopenia | 7 (25.0) | 1 (3.6) |
| Fall | 6 (21.4) | 3 (10.7) |
| Cellulitis | 4 (14.3) | 2 (7.1) |
| Acute kidney injury | 3 (10.7) | 2 (7.1) |
| Pneumonia | 3 (10.7) | 3 (10.7) |
| Hypertension | 2 (7.1) | 1 (3.6) |
| Syncope | 2 (7.1) | 2 (7.1) |
All-grade cardiac TEAEs included palpitations (n = 2 [7%]) and arrhythmia, atrial fibrillation, atrial flutter, and congestive cardiac failure (n = 1 each [4%]; atrial fibrillation and cardiac failure were considered treatment-related). Congestive cardiac failure was the only grade 3/4 cardiac TEAE. All-grade renal/urinary disorder TEAEs included pollakiuria (n = 4 [14%]), acute kidney injury (n = 3 [11%]; 1 [4%] treatment-related), hematuria, renal impairment, urinary retention (n = 2 each [7%]; 1 [4%] renal impairment considered treatment-related), and chronic kidney disease, dysuria, nephrolithiasis, nocturia, urinary incontinence, and urinary tract pain (n = 1 each [4%]; incontinence and pain were considered treatment-related). The only grade 3/4 renal/urinary disorder TEAE was acute kidney injury (n = 2 [7%]).
Serious TEAEs occurred in 12 (43%) patients and included fall and acute kidney injury (11% each) and pneumonia and cellulitis (7% each; cellulitis not related to injection site). A total of 5 (18%) patients died: 3 (11%) because of complications of high-dose melphalan and ASCT (septic shock and multiorgan system failure, recurrent infections, and septic shock, respectively), and 2 (7%) because of progression of amyloidosis-related organ dysfunction.
An IRR occurred in 1 (4%) patient, comprising chest discomfort, cough, hypotension, oropharyngeal pain, and sneezing, all of which were grade 1. All occurred on cycle 1 day 1 except hypotension (cycle 1, day 8; considered probably related to DARA SC), and all resolved. A total of 6 injection-site reactions occurred in 3 (11%) patients. All injection-site reactions were grade 1 and included erythema, bruising, and skin discoloration; none led to changes in treatment.
Efficacy
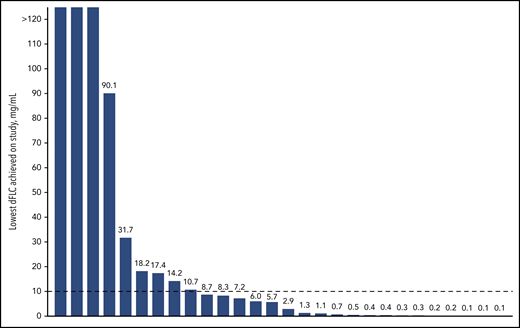
The ORR (best response) to therapy with DARA SC plus CyBorD was 96% at a median follow-up of 17.6 months. A total of 15 (54%) patients achieved CR or mCR; 10 (36%) patients achieved CR based on consensus criteria, and 5 (18%) patients achieved CR based on all criteria except normalization of the FLC ratio (mCR; Figure 2A; supplemental Appendix, available on the Blood Web site). Twenty-three (82%) patients achieved VGPR or better. PR or better was achieved by 20 (71%) patients at 1 month, 22 (79%) patients at 3 months, and 17 (61%) patients at 6 months. The number of patients achieving deep hematologic responses as measured by dFLC lower than 10 mg/L44 and iFLC of 20 mg/L or less43 were 19 (68%; Figure 3) and 20 (71%), respectively.
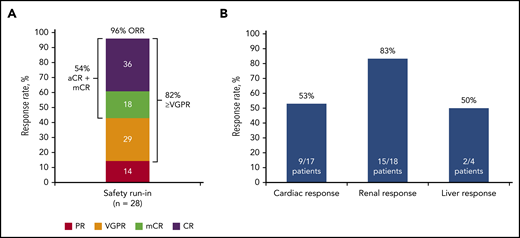
Summary of overall best hematologic and organ responses. Overall best response for hematologic responses (A) and organ responses (B). Patients who met VGPR criteria and also had negative serum and urine immunofixation and normalization of iFLC, but with uFLC below the lower limit of normal (FLC ratio abnormal or normal) who therefore did not meet the criteria for CR were included in the mCR group.
Summary of overall best hematologic and organ responses. Overall best response for hematologic responses (A) and organ responses (B). Patients who met VGPR criteria and also had negative serum and urine immunofixation and normalization of iFLC, but with uFLC below the lower limit of normal (FLC ratio abnormal or normal) who therefore did not meet the criteria for CR were included in the mCR group.
In responders, the median time to first response (PR or better) was 9 (range, 7-85) days, median time to VGPR was 19 (range, 7-339) days, and median time to aCR+mCR was 85 (range, 29-179) days. The median duration of aCR+mCR has not been reached, and responses deepened with time (Figure 1). At a median follow-up of 17.6 months, all patients achieving aCR+mCR remained in hematologic remission (only 1 patient underwent ASCT for consolidation of CR). Among 9 patients who proceeded to ASCT, only 1 was in CR before transplant. Of the 7 patients with measurable organ involvement at baseline, 5 (71%) patients had an organ response before ASCT (2 cardiac and 3 renal). There are currently 6 patients evaluable for post-ASCT hematologic response, of whom 4 (67%) deepened their response. Three of 9 patients died of transplant-related complications; their clinical courses are summarized in the supplemental Appendix.
The overall organ response rate (any evaluable organ; heart, kidney, and/or liver) was 64% at a median follow-up of 17.6 months. Responses for specific organs are shown in Figure 2B. In patients with cardiac involvement, responses were seen in 9 (53%) of 17 patients, with 6 (38%) of 16 evaluable patients having a response at 3 months, 6 (46%) of 13 evaluable patients having a response at 6 months, and 8 (62%) of 13 evaluable patients with a response at 12 months. Among those with renal involvement, responses were observed in 15 (83%) of 18 patients: in 6 (38%) of 16 evaluable patients at 3 months, 7 (47%) of 15 evaluable patients at 6 months, and 10 (67%) of 15 patients at 12 months. Among the 4 patients with hepatic involvement, 2 showed a response. In these patients, a response was seen in 0 of 2 evaluable patients at 3 months, 0 of 3 evaluable patients at 6 months, and 2 of 3 evaluable patients at 12 months. The median time to response in cardiac responders was 114 (range, 29-561) days; for renal responders, it was 57 (range, 29-449) days, and for hepatic responders, it was 330 (range, 321-338) days.
Discussion
In the safety run-in cohort of the phase 3 ANDROMEDA study, DARA SC plus CyBorD was well tolerated in patients with previously untreated AL amyloidosis. No new safety concerns were identified with DARA SC plus CyBorD compared with daratumumab monotherapy (IV or SC) or CyBorD alone.46-50 DARA SC is associated with low rates of IRRs, few injection-site reactions, and reduced administration times compared with DARA IV.48 The advantages of DARA SC as reported in the phase 3 COLUMBA study in MM,48 particularly the small administration volume, are relevant to patients with AL amyloidosis for whom volume overload is a concern because of cardiac involvement. The safety profile of DARA SC plus CyBorD compared with that of CyBorD alone will be examined further in the randomized portion of the ANDROMEDA study.
The depth and rapidity of hematologic response to DARA SC plus CyBorD were notable. In addition, the majority of patients achieved an absolute dFLC level lower than 10 mg/L or an iFLC level lower than 20 mg/L.43,44 These stringent hematologic responses induced substantial organ responses; an overall organ response rate of 64% for the heart, kidney, and/or liver demonstrates clinically relevant functional improvement in organs most frequently affected by AL amyloidosis.
Criteria for defining hematologic response in AL amyloidosis have evolved in parallel with diagnostic and therapeutic advances37 (Table 3). Development of assays to detect serum FLC levels allows measurement of hematologic responses in most patients, and the absolute depth of FLC response is now known to affect patient outcomes.43,44 Although negative serum and urine IFE remain requirements for achievement of aCR, investigators have begun to consider the absolute dFLC or iFLC levels a more relevant measure of hematologic response. Importantly, Muchtar et al demonstrated that normalization of FLC has no effect on OS or organ response rate compared with an abnormal FLC ratio. These investigators also showed that an absolute iFLC level of 20 mg/L or less is associated with improved survival.43 Manwani et al recently reported outcomes of patients with newly diagnosed AL (N = 915) treated with bortezomib-based therapy (95% received CyBorD), using a dFLC level lower than 10 mg/L as a “stringent dFLC response.” Achieving a stringent dFLC response was associated with improved OS and time to next treatment compared with less deep responses. Cardiac and renal responses were significantly higher among those achieving a stringent dFLC response.44 These findings strongly support the concept that the absolute reduction of the amyloidogenic light chain is the most physiologically relevant measure of hematologic response and outcomes in AL amyloidosis, and that the current international consensus criteria should be re-evaluated. An additional methodologic advance to assess and manage patients with AL amyloidosis is evaluation of minimal residual disease, as the absence of minimal residual disease may be associated with deeper organ response.51
Definitions of deep hematologic responses in AL amyloidosis are evolving
| Parameter | Complete response | Modified complete response* | Stringent dFLC response* | Absolute iFLC response* | ||
|---|---|---|---|---|---|---|
| Gertz 200537 | Comenzo 201214 | Palladini 201253 | AMY3001 2020 | Manwani 201944 | Muchtar 201943 | |
| Negative serum IFE | ✓ | ✓ | ✓ | ✓ | Not required | Not required |
| Negative urine IFE | ✓ | ✓ | ✓ | ✓ | Not required | Not required |
| Bone marrow plasma cells <5% | ✓ | Not required | Not required | Not required | Not required | Not required |
| FLC | Normal levels | Normal levels | Not required | iFLC <ULN | dFLC <10 mg/L | iFLC ≤20 mg/L |
| FLC ratio (normal) | ✓ | ✓ | ✓ | Not required | Not required | Not required |
| Parameter | Complete response | Modified complete response* | Stringent dFLC response* | Absolute iFLC response* | ||
|---|---|---|---|---|---|---|
| Gertz 200537 | Comenzo 201214 | Palladini 201253 | AMY3001 2020 | Manwani 201944 | Muchtar 201943 | |
| Negative serum IFE | ✓ | ✓ | ✓ | ✓ | Not required | Not required |
| Negative urine IFE | ✓ | ✓ | ✓ | ✓ | Not required | Not required |
| Bone marrow plasma cells <5% | ✓ | Not required | Not required | Not required | Not required | Not required |
| FLC | Normal levels | Normal levels | Not required | iFLC <ULN | dFLC <10 mg/L | iFLC ≤20 mg/L |
| FLC ratio (normal) | ✓ | ✓ | ✓ | Not required | Not required | Not required |
Pending validation and international consensus agreement.
In this trial, we assessed 5 patients as achieving mCR, defined as a negative serum and urine IFE and iFLC less than the upper limit of normal, regardless of the uFLC level or FLC ratio. Of the 4 patients reaching mCR who had baseline cardiac, renal, or hepatic involvement, 3 achieved organ responses for each of the respective involved organs; the fourth achieved a response for 1 of 4 involved organs. More than half of patients in the ANDROMEDA safety run-in cohort achieved either aCR or mCR. Notably, these promising hematologic responses were durable, as all 15 patients achieving aCR+mCR remained in remission at a median follow-up of 17.6 months.
No therapies have been approved for AL amyloidosis. At this time, the most commonly used front-line regimens for this disease are CyBorD,7 melphalan with dexamethasone,52 and high-dose melphalan (HDM) with ASCT, although the latter is not feasible for many patients with AL amyloidosis.9-11 In patients with newly diagnosed AL amyloidosis receiving CyBorD,7 melphalan with dexamethasone,52 and HDM with ASCT,9-11 hematologic CR rates were 23%, 12%, and 34% to 48%, respectively. DARA SC plus CyBorD achieved a stringent dFLC response in 68% of patients compared with 30% with bortezomib-based combinations (95% received CyBorD).44 Hematologic responses in the safety run-in cohort of ANDROMEDA compare favorably to all commonly used frontline regimens, including HDM and ASCT. Cardiac and renal response rates in the ANDROMEDA safety run-in cohort were 53% and 83%, respectively, and compare favorably with the current standard nontransplant regimens presented here. Respective cardiac and renal response rates were 17% and 25% for CyBorD, and 20% and 17% for melphalan with dexamethasone.7,52 Among patients who achieved CR with HDM and ASCT, the organ response rate was 79%.9
In summary, this is the first report on the use of daratumumab in combination with CyBorD in patients with newly diagnosed AL amyloidosis, and the first report of DARA SC treatment of a disease other than MM. DARA SC plus CyBorD was well tolerated in the safety run-in portion of the phase 3 ANDROMEDA study. No new safety concerns were identified compared with intravenous or subcutaneous daratumumab monotherapy or CyBorD alone, and low rates of IRRs were observed. Preliminary efficacy was robust, with high rates of deep and durable hematologic responses, and importantly, organ responses elicited by DARA SC plus CyBorD. These results indicate that DARA SC plus CyBorD is a promising treatment of AL amyloidosis, and support the ongoing randomized portion of ANDROMEDA.
The data sharing policy of Janssen Pharmaceutical Companies of Johnson & Johnson is available at https://www.janssen.com/clinical-trials/transparency. As noted on this site, requests for access to the study data can be submitted through the Yale Open Data Access Project site at http://yoda.yale.edu.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the patients who participated in the ANDROMEDA study and their families, study investigators, and staff at each of the clinical sites; staff involved in data collection and analyses; and members of the Data and Safety Monitoring Committee.
This study was sponsored by Janssen Research & Development, LLC. Medical writing and editorial support were provided by Elise Blankenship and Jill E. Kolesar of MedErgy, and were funded by Janssen Global Services, LLC.
Authorship
Contribution: All authors drafted and reviewed the manuscript, approved the final version, decided to publish this report, and vouch for data accuracy and completeness.
Conflict-of-interest disclosure: G.P. received an honorarium from Sebia, served on an advisory board for Janssen, and received a travel grant from Celgene. E.K. received honoraria from Amgen, Genesis Pharma, Janssen, Takeda, and Prothena, and received research funding from Amgen and Janssen. M.S.M. served in a consulting or advisory role for Pfizer, Akcea, Eldos, Ionis, and Prothena and received research funding from Pfizer, Eldos, and Alnylam. J.Z. served in a consulting or advisory role for Takeda, Janssen, Celgene, Bristol-Myers Squibb, Prothena, Caelum, Alnylam, and Amgen and received research support from Celgene. M.C.M. served in a consulting or advisory role for Janssen Cilag, Celgene, Servier, Gilead, and Amgen and received research funding from Celgene. A.D.W. received honoraria from Janssen, Celgene, Prothena, and Takeda; served in a consulting or advisory role for GlaxoSmithKline and Karyopharm; and received research funding from Amgen. A.J. served in a consulting or advisory role for Janssen and received honoraria from, received research funding from, and had travel, accommodations, or other expenses paid or reimbursed by Janssen and Celgene. H.C.L. served in a consulting or advisory role for and received research funding from Amgen, Celgene, GlaxoSmithKline, Janssen, and Takeda; served in a consulting or advisory role for Sanofi; and received research funding from Daiichi Sankyo. J.L.K. consulted for AbbVie, Roche, Takeda, Janssen, and Pharmacyclics and received research funding from Amgen and Novartis. T.K. served in a consulting or advisory role for Celgene and Amgen and received research support from AbbVie, Amgen, Janssen, and Prothena. M.R. served on a speakers bureau for Janssen, Akcea, Celgene, and Takeda. V.S. served in a consulting or advisory role for Caelum and Proclara and received research funding (to institution) from Janssen, Takeda, Celgene, and Prothena. R.L.C. served in a consulting or advisory role for Janssen, Karyopharm, Takeda, Sanofi-Aventis, Caelum, and Prothena; has an immediate family member holding patents, patents pending, received royalties, participated in royalty sharing agreements, or other intellectual property interests from a discovery or technology related to health or medicine for Janssen; and received research funding from Janssen. X.Q., S.Y.V., B.M.W., and J.V. are employees of Janssen. The remaining authors declare no competing financial interests.
Correspondence: Giovanni Palladini, Amyloidosis Research and Treatment Center, Fondazione Istituto di Ricovero e Cura a Carattere Scientifico Policlinico San Matteo, Department of Molecular Medicine, University of Pavia, 6, Via C Forlanini, 27100, Pavia, Italy; e-mail: giovanni.palladini@unipv.it.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal