

Key Points
Acalabrutinib is clinically effective in relapsed/refractory and high-risk treatment naïve chronic lymphocytic leukemia.
BID dosing maintains near complete occupancy of BTK in blood and tissues and more profoundly inhibits oncogenic signaling than QD dosing.

Abstract
Inhibition of the B-cell receptor pathway, and specifically of Bruton tyrosine kinase (BTK), is a leading therapeutic strategy in B-cell malignancies, including chronic lymphocytic leukemia (CLL). Target occupancy is a measure of covalent binding to BTK and has been applied as a pharmacodynamic parameter in clinical studies of BTK inhibitors. However, the kinetics of de novo BTK synthesis, which determines occupancy, and the relationship between occupancy, pathway inhibition and clinical outcomes remain undefined. This randomized phase 2 study investigated the safety, efficacy, and pharmacodynamics of a selective BTK inhibitor acalabrutinib at 100 mg twice daily (BID) or 200 mg once daily (QD) in 48 patients with relapsed/refractory or high-risk treatment-naïve CLL. Acalabrutinib was well tolerated and yielded an overall response rate (ORR) of partial response or better of 95.8% (95% confidence interval [CI], 78.9-99.9) and an estimated progression-free survival (PFS) rate at 24 months of 91.5% (95% CI, 70.0-97.8) with BID dosing and an ORR of 79.2% (95% CI, 57.9-92.9) and an estimated PFS rate at 24 months of 87.2% (95% CI, 57.2-96.7) with QD dosing. BTK resynthesis was faster in patients with CLL than in healthy volunteers. BID dosing maintained higher BTK occupancy and achieved more potent pathway inhibition compared with QD dosing. Small increments in occupancy attained by BID dosing relative to QD dosing compounded over time to augment downstream biological effects. The impact of BTK occupancy on long-term clinical outcomes remains to be determined. This trial was registered at www.clinicaltrials.gov as #NCT02337829.
Introduction
Bruton tyrosine kinase (BTK) is a proximal component of the B-cell receptor (BCR) signaling pathway and a validated target for B-cell malignancies, including chronic lymphocytic leukemia (CLL). BTK is irreversibly inhibited by acalabrutinib, which forms a covalent bond with Cys481 in the ATP-binding pocket. In contrast to the first-in-class BTK inhibitor ibrutinib, acalabrutinib is very selective and does not inhibit other kinases with the conserved cysteine residue, including EGFR, TEC, or ITK.1,2 Inhibition of both target and nontarget kinases is thought to contribute to ibrutinib-related adverse events such as bleeding diathesis via inhibition of BTK and TEC3,4 and diarrhea via inhibition of EGFR.5 Toxicity is among the most common reasons for discontinuation of ibrutinib.6 For patients intolerant to ibrutinib, treatment with acalabrutinib is generally well tolerated.7
Covalent binding to BTK prolongs the pharmacodynamic effects of acalabrutinib despite a short pharmacokinetic half-life.1 The amount of BTK occupied by acalabrutinib is correlated with inhibition of BCR signaling in healthy volunteers8 and has been used as a pharmacodynamic endpoint in clinical studies. Although most studies to date have reported BTK occupancy in circulating CLL cells, activation of the BCR primarily occurs in the lymph node.9 In addition to supporting survival and proliferation,9 BCR signaling drives global messenger RNA (mRNA) translation in CLL cells.10 Consequently, the rate of BTK resynthesis may differ between blood and lymph node. In a phase 1 study of a different BTK inhibitor zanubrutinib, BTK occupancy in lymph node was more variable than in blood.11 Understanding the correlation of BTK occupancy between anatomic compartments is needed to infer pharmacologic activity in tissues from peripheral blood measurements.
In a phase 1-2 study of acalabrutinib for relapsed CLL, near complete target occupancy in peripheral blood was better maintained with twice daily (BID) compared with once daily (QD) dosing during the first week of treatment.1 Median BTK occupancy at the time of drug trough exposure was 97% in patients receiving acalabrutinib 100 mg BID group and 92% in those receiving 250 mg QD. Median peak BTK occupancy was 99% in both groups. BID administration has been used in subsequent clinical studies of acalabrutinib and is the approved dosing regimen for the treatment of CLL or small lymphocytic lymphoma (SLL) and relapsed/refractory mantle cell lymphoma in the United States.12 Nevertheless, it is unknown whether fluctuations in BTK occupancy affect biological readouts of BCR pathway inhibition and clinical outcomes.
We conducted a phase 2 study of acalabrutinib 100 mg BID or 200 mg QD in patients with relapsed/refractory or high-risk treatment-naïve CLL. Correlative studies investigating the spatial and temporal features of BTK turnover were emphasized in the study design. Efficacy, safety, and pharmacodynamic analyses of this study are reported herein.
Materials and methods
Study design and participants
This is a phase 2 open-label, single-center study of acalabrutinib 100 mg BID or 200 mg QD. Eligibility criteria included active CLL or SLL requiring treatment as defined by the International Workshop on Chronic Lymphocytic Leukemia (IWCLL)13 ; relapsed or refractory CLL/SLL or treatment-naïve patients with 17p deletion, TP53 mutation, or NOTCH1 mutation; absolute neutrophil count >750/μL; platelet count >30 000/μL; an Eastern Cooperative Oncology Group performance status ≤2; adequate organ function, defined by estimated glomerular filtration rate >50, total bilirubin <1.5 × the upper limit of normal, alanine aminotransferase, and aspartate aminotransferase <2.5 × upper limit of normal. Exclusion criteria included Richter transformation, prior BTK inhibitor therapy; autoimmune cytopenia requiring corticosteroid therapy; life-threatening illness; any cancer with expected survival of <2 years, warfarin anticoagulation; malabsorption disorder; and uncontrolled or symptomatic cardiovascular disease. The study was approved by the National Heart, Lung, and Blood Institute Institutional Review Board. All patients provided written informed consent.
Evaluation, randomization, and treatment
Baseline assessment included IGHV sequencing, interphase cytogenetic analysis, TP53 and NOTCH1 sequencing, bone marrow biopsy, and computed tomography (CT) of the neck, chest, abdomen, and pelvis. Lymph node core needle biopsies or bone marrow biopsies were obtained in patients with or without superficial lymphadenopathy, respectively, before and after the first 3 days of treatment. Block randomization was used to assign patients within each biopsy group to acalabrutinib 100 mg BID or 200 mg QD. Acalabrutinib was administered in 28-day cycles as tolerated until disease progression. The study was amended in May 2018 to permit patients receiving acalabrutinib 200 mg QD to switch to 100 mg BID after the first 6 cycles of treatment. Acalabrutinib was withheld for first and second occurrences of grade 4 neutropenia and thrombocytopenia and grade ≥ 3 non-hematologic toxicities then resumed at the original dose after resolution, dose-reduced to 100 mg once daily after a third occurrence, and permanently discontinued after a fourth occurrence. Patients were evaluated at baseline, monthly for 6 months, then every 3 months thereafter with history, physical examination, and laboratory testing.
Outcomes
The primary endpoint was best response to treatment in the combined BID and QD groups based on the IWCLL criteria13 incorporating clarification for response assessment in patients treated with kinase inhibitors.14 Secondary endpoints included safety and tolerability, progression-free and overall survival, BTK occupancy, and characterization of tumor biology on treatment. Response, including CT, was assessed after 2 cycles, 6 cycles, and annually. Bone marrow biopsy was performed after 6 cycles, 12 cycles, and to confirm complete responses. Common Terminology Criteria for Adverse Events, version 4.03, and the IWCLL guidelines13 were used to grade the severity of nonhematologic and hematologic adverse events, respectively.
Statistical analysis
A sample size of 44 was calculated to provide 85% power and a type I error of 0.05 to reject an overall response rate (ORR) of ≤50% if the ORR was ≥70%. Descriptive statistics were used to summarize findings. ORR and 95% confidence interval (CI) were estimated using sample proportions based on binomial distribution. Progression-free survival (PFS) was estimated by the Kaplan-Meier method and compared between subgroups by the log-rank test. Data are presented through December 7, 2018. Patients without an event were censored at the time of last clinical assessment.
Flow cytometry
Disease burden was quantified by flow cytometry according to methods recommended by the European Research Initiative in CLL.15 Minimal residual disease negativity was defined as fewer than 1 CLL cell in 10 000 leukocytes (<10−4) assessed.
BTK and PLCG2 sequencing
Tumor cells were isolated from peripheral blood with Dynabeads CD19 Pan B (Invitrogen, Waltham, MA) followed by DNA extraction with QIAamp DNA Blood Mini Kit (Qiagen, Germantown, MD). Exonic and flanking regions of 10 putative driver genes, including BTK, were amplified using a TruSeq Custom Amplicon Panel (Illumina, San Diego, CA) then sequenced on the MiSeq System (Illumina). Sequencing reads were aligned to the human genome build 19 (hg19) and annotated using Illumina VariantStudio. Variants were identified with detection thresholds of 500× coverage and variant allele frequency >5%.
PLCG2 exons 19, 20, and 24 were amplified using custom oligonucleotides and analyzed by bidirectional Sanger sequencing (NeoGenomics, Fort Myers, FL). A high-sensitivity assay base on wild-type blocking polymerase chain reactions with branched or locked nucleic acids was performed to detect mutations with variant allele frequency as low as 0.1% (NeoGenomics).16
BTK occupancy
Occupancy of BTK by acalabrutinib was measured in cryopreserved peripheral blood mononuclear cells, lymph node, and bone marrow samples with the aid of a biotin-tagged analog probe using an enzyme-linked immunosorbent assay-based method as previously described.1 Peripheral blood samples were collected from all patients at baseline, during a dose interruption from day 3 to 5 to measure the rate of BTK resynthesis (at 4, 12, 24, 36, and 48 hours after administration of acalabrutinib), and before administration of acalabrutinib after 1 cycle, 6 cycles, and 12 cycles of treatment. For patients who switched from 200 mg QD to 100 mg BID after the first 6 cycles, additional samples were collected before and after the dose switch for BTK occupancy analysis.
RNA sequencing and analysis
RNA sequencing (RNA-seq) libraries were prepared using the TruSeq Stranded total RNA Library Preparation Gold Kit (Illumina) and sequenced on a HiSeq 3000 System. Paired gene-level differential analysis was conducted using limma.17 Limma-voom17,18 was used to implement a gene-wise linear modeling which processes the read counts into log2CPM with associated precision weights. Log2CPM values were normalized between samples using trimmed mean of M-values.19 Preranked gene set enrichment analysis was performed20 using a curated set of lymphocyte gene-expression signatures (supplemental Table 1, available on the Blood Web site).21 Details are provided in supplemental Methods.
Results
Study population
Between January 13, 2015, and June 15, 2018, 48 patients were enrolled and randomized to receive acalabrutinib 100 mg BID or 200 mg QD (Figure 1). The median age was 64.0 years (range, 45-83) and 16 (33%) patients were treatment naϊve. 37.5% of patients had bulky lymphadenopathy (≥5 cm), 37.5% had advanced Rai stage disease, 77.1% had unmutated IGHV, 32.5% had del 17p and/or TP53 mutation, and 45.2% had NOTCH1 mutation (Table 1). Baseline disease characteristics were not significantly different between the BID and QD groups (P > .05, Fisher’s exact test), although the number of patients with TP53 aberration was numerically higher in the QD group (42.9%) than the BID group (21.1%). Among 32 patients with relapsed/refractory (R/R) CLL, the median number of prior therapies was 1 (range 1-2; supplemental Table 2).
Patient characteristics at baseline
| 100 mg BID (n = 24) (%) | 200 mg QD (n = 24) (%) | Total (n = 48) (%) | |
|---|---|---|---|
| Disease status | |||
| Treatment-naive | 6 (25.0) | 10 (41.7) | 16 (33.3) |
| R/R | 18 (75.0) | 14 (58.3) | 32 (66.7) |
| Histology | |||
| CLL | 22 (91.7) | 21 (87.5) | 43 (89.6) |
| SLL | 2 (8.3) | 3 (12.5) | 5 (10.4) |
| Baseline Rai stage | |||
| 0 (low risk) | 0 | 0 | 0 |
| I-II (intermediate risk) | 15 (62.5) | 11 (45.8) | 26 (54.2) |
| III-IV (high risk) | 7 (29.2) | 11 (45.8) | 18 (37.5) |
| Missing | 2 (8.3) | 2 (8.3) | 4 (8.3) |
| Bulky disease | |||
| ≥5 cm | 11 (45.8) | 7 (29.2) | 18 (37.5) |
| Genetic aberrations | |||
| Del 17p/TP53 mutation | 4/19 (21.1) | 9/21 (42.9) | 13/40 (32.5) |
| Del 17p only | 1/20 (5.0) | 1/21 (4.8) | 2/41 (4.9) |
| TP53 mutation only | 2/20 (10.0) | 3/22 (13.6) | 5/42 (11.9) |
| Both del 17p and TP53 mutation | 1/19 (5.3) | 5/21 (23.8) | 6/40 (15) |
| Del 11q | 4/20 (20.0) | 5/21 (23.8) | 9/41 (22.0) |
| NOTCH1 mutation | 8/20 (40.0) | 11/22 (50.0) | 19/42 (45.2) |
| IGHV | |||
| Unmutated | 17 (70.8) | 20 (83.3) | 37 (77.1) |
| Mutated | 3 (12.5) | 2 (8.3) | 5 (10.4) |
| B2-microglobulin | |||
| >3.5 mg/L | 11 (45.8) | 9 (37.5) | 20 (41.7) |
| 100 mg BID (n = 24) (%) | 200 mg QD (n = 24) (%) | Total (n = 48) (%) | |
|---|---|---|---|
| Disease status | |||
| Treatment-naive | 6 (25.0) | 10 (41.7) | 16 (33.3) |
| R/R | 18 (75.0) | 14 (58.3) | 32 (66.7) |
| Histology | |||
| CLL | 22 (91.7) | 21 (87.5) | 43 (89.6) |
| SLL | 2 (8.3) | 3 (12.5) | 5 (10.4) |
| Baseline Rai stage | |||
| 0 (low risk) | 0 | 0 | 0 |
| I-II (intermediate risk) | 15 (62.5) | 11 (45.8) | 26 (54.2) |
| III-IV (high risk) | 7 (29.2) | 11 (45.8) | 18 (37.5) |
| Missing | 2 (8.3) | 2 (8.3) | 4 (8.3) |
| Bulky disease | |||
| ≥5 cm | 11 (45.8) | 7 (29.2) | 18 (37.5) |
| Genetic aberrations | |||
| Del 17p/TP53 mutation | 4/19 (21.1) | 9/21 (42.9) | 13/40 (32.5) |
| Del 17p only | 1/20 (5.0) | 1/21 (4.8) | 2/41 (4.9) |
| TP53 mutation only | 2/20 (10.0) | 3/22 (13.6) | 5/42 (11.9) |
| Both del 17p and TP53 mutation | 1/19 (5.3) | 5/21 (23.8) | 6/40 (15) |
| Del 11q | 4/20 (20.0) | 5/21 (23.8) | 9/41 (22.0) |
| NOTCH1 mutation | 8/20 (40.0) | 11/22 (50.0) | 19/42 (45.2) |
| IGHV | |||
| Unmutated | 17 (70.8) | 20 (83.3) | 37 (77.1) |
| Mutated | 3 (12.5) | 2 (8.3) | 5 (10.4) |
| B2-microglobulin | |||
| >3.5 mg/L | 11 (45.8) | 9 (37.5) | 20 (41.7) |
The median time on treatment was 25.5 months (range, 1.8-46.4) for the entire study, 26.1 months (range, 5.7-46.4) for the BID group, and 25.5 months (range, 1.8-46.0) for the QD group. The median relative dose intensity was 98.7% (range, 73.0%-101.3%) for the BID group and 98.7% (range, 75.8%-99.8%) for the QD group. Twelve patients have discontinued treatment, most commonly for adverse events (n = 6) or progressive disease (n = 4) (supplemental Table 3).
Efficacy
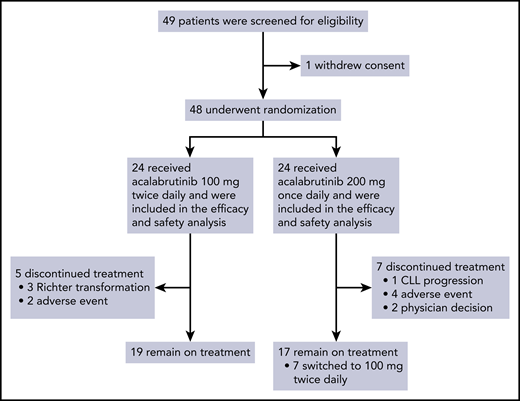
Efficacy was assessed in the intent-to-treat population of 48 patients who underwent randomization. The median time to initial partial response or better was 5.5 months (range, 0.9-23.9) for the BID group, and 5.5 months (range, 1.8-35.9) for the QD group. The ORR was 87.5% (95% CI, 74.8-95.3). The ORR was 95.8% (95% CI, 78.9-99.9) in the BID group and 79.2% (95% CI, 57.9-92.9) in the QD group (Figure 2A). Age, Rai stage, prior treatment, del 17p and/or TP53 mutation, and NOTCH1 mutation were not associated with a difference in ORR (Table 2).
Efficacy of acalabrutinib. (A) The rate of overall response, including complete remission and partial remission, in the BID group and the QD group. (B) Absolute lymphocyte count during treatment. (C) Maximum percent change in the sum of the product of dimensions during treatment. (D) Quantification of disease burden in bone marrow and peripheral blood separated by dosing groups. Line denotes median of each group at the indicated time point. *P < .05. NED, no evidence of disease.
Efficacy of acalabrutinib. (A) The rate of overall response, including complete remission and partial remission, in the BID group and the QD group. (B) Absolute lymphocyte count during treatment. (C) Maximum percent change in the sum of the product of dimensions during treatment. (D) Quantification of disease burden in bone marrow and peripheral blood separated by dosing groups. Line denotes median of each group at the indicated time point. *P < .05. NED, no evidence of disease.
Response rates to acalabrutinib
| 100 mg BID (n = 24) (%) | 200 mg QD (n = 24) (%) | Total (n = 48) (%) | |
|---|---|---|---|
| ORR | 23 (95.8) | 19 (79.2) | 42 (87.5) |
| 95% CI | (78.9-99.9) | (57.9-92.9) | (74.8-95.3) |
| CR | 1 (4.2) | 3 (12.5) | 4 (8.3) |
| PR | 22 (91.7) | 16 (66.7) | 38 (79.2) |
| PRL | 0 | 0 | 0 |
| Age, y | |||
| <65 | 11/11 (100.0) | 14/16 (87.5) | 25/27 (92.6) |
| (71.5-100.0) | (61.7-98.5) | (75.7-99.1) | |
| ≥65 | 12/13 (92.3) | 5/8 (62.5) | 17/21 (81.0) |
| (64.0-99.8) | (24.5-91.5) | (58.1-94.6) | |
| Rai stage | |||
| Intermediate | 14/14 (100.0) | 10/11 (90.9) | 24/25 (96.0) |
| (76.8-100.0) | (58.7-99.8) | (79.7-99.9) | |
| High | 7/8 (87.5) | 7/11 (63.6) | 14/19 (73.7) |
| (47.4-99.7) | (30.8-89.1) | (48.8-90.9) | |
| Prior treatment | |||
| No | 6/6 (100.0) | 8/10 (80.0) | 14/16 (87.5) |
| (54.1-100.0) | (44.4-97.5) | (61.7-98.5) | |
| Yes | 17/18 (94.4) | 11/14 (78.6) | 28/32 (87.5) |
| (72.7-99.9) | (49.2-95.3) | (71.0-96.5) | |
| Del 17p/TP53 mutation | |||
| No | 14/15 (93.3) | 11/12 (91.7) | 25/27 (92.6) |
| (68.1-99.8) | (61.5-99.8) | (75.7-99.1) | |
| Yes | 4/4 (100.0) | 6/9 (66.7) | 10/13 (76.9) |
| (39.8-100.0) | (29.9-92.5) | (46.2-95.0) | |
| NOTCH1 mutation | |||
| No | 11/12 (91.7) | 8/11 (72.7) | 19/23 (82.6) |
| (61.5-99.8) | (39.0-94.0) | (61.2-95.1) | |
| Yes | 8/8 (100.0) | 10/11 (90.9) | 18/19 (94.7) |
| (63.1-100.0) | (58.7-99.8) | (74.0-99.9) |
| 100 mg BID (n = 24) (%) | 200 mg QD (n = 24) (%) | Total (n = 48) (%) | |
|---|---|---|---|
| ORR | 23 (95.8) | 19 (79.2) | 42 (87.5) |
| 95% CI | (78.9-99.9) | (57.9-92.9) | (74.8-95.3) |
| CR | 1 (4.2) | 3 (12.5) | 4 (8.3) |
| PR | 22 (91.7) | 16 (66.7) | 38 (79.2) |
| PRL | 0 | 0 | 0 |
| Age, y | |||
| <65 | 11/11 (100.0) | 14/16 (87.5) | 25/27 (92.6) |
| (71.5-100.0) | (61.7-98.5) | (75.7-99.1) | |
| ≥65 | 12/13 (92.3) | 5/8 (62.5) | 17/21 (81.0) |
| (64.0-99.8) | (24.5-91.5) | (58.1-94.6) | |
| Rai stage | |||
| Intermediate | 14/14 (100.0) | 10/11 (90.9) | 24/25 (96.0) |
| (76.8-100.0) | (58.7-99.8) | (79.7-99.9) | |
| High | 7/8 (87.5) | 7/11 (63.6) | 14/19 (73.7) |
| (47.4-99.7) | (30.8-89.1) | (48.8-90.9) | |
| Prior treatment | |||
| No | 6/6 (100.0) | 8/10 (80.0) | 14/16 (87.5) |
| (54.1-100.0) | (44.4-97.5) | (61.7-98.5) | |
| Yes | 17/18 (94.4) | 11/14 (78.6) | 28/32 (87.5) |
| (72.7-99.9) | (49.2-95.3) | (71.0-96.5) | |
| Del 17p/TP53 mutation | |||
| No | 14/15 (93.3) | 11/12 (91.7) | 25/27 (92.6) |
| (68.1-99.8) | (61.5-99.8) | (75.7-99.1) | |
| Yes | 4/4 (100.0) | 6/9 (66.7) | 10/13 (76.9) |
| (39.8-100.0) | (29.9-92.5) | (46.2-95.0) | |
| NOTCH1 mutation | |||
| No | 11/12 (91.7) | 8/11 (72.7) | 19/23 (82.6) |
| (61.5-99.8) | (39.0-94.0) | (61.2-95.1) | |
| Yes | 8/8 (100.0) | 10/11 (90.9) | 18/19 (94.7) |
| (63.1-100.0) | (58.7-99.8) | (74.0-99.9) |
CR, complete response; PR, partial response; PRL, partial response with lymphocytosis.
Treatment-related lymphocytosis was observed in 36 (66.7%) patients. The median time to lymphocytosis was 0.4 weeks (range, 0.4-7.9) and the median duration of lymphocytosis was 8.4 weeks (95% CI, 3.7-11.7). The median absolute lymphocyte count was 6.52 K/μL (95% CI, 5.28-11.05) after 12 cycles and 3.09 K/μL (95% CI, 1.36-5.23) after 24 cycles (Figure 2B). Reduction in lymphadenopathy was detected early and improved with extended treatment. The median time to first nodal response was 7.9 weeks (range, 7.4-26.7), which coincided with first CT evaluation after treatment initiation. At best response, 38 (69%) patients still had ≥1 lymph node ≥1.5 cm in the greatest diameter (Figure 2C). Disease burden in blood and bone marrow, as quantified by flow cytometry, decreased with extended treatment (Figure 2D). The BID group had less residual disease in the marrow than the QD group after 6 cycles (P = .039) and 12 cycles (P = .034) of treatment. One patient in the BID group achieved minimal residual disease negativity (<10−4) in blood and bone marrow after 6 cycles and 12 cycles, respectively.
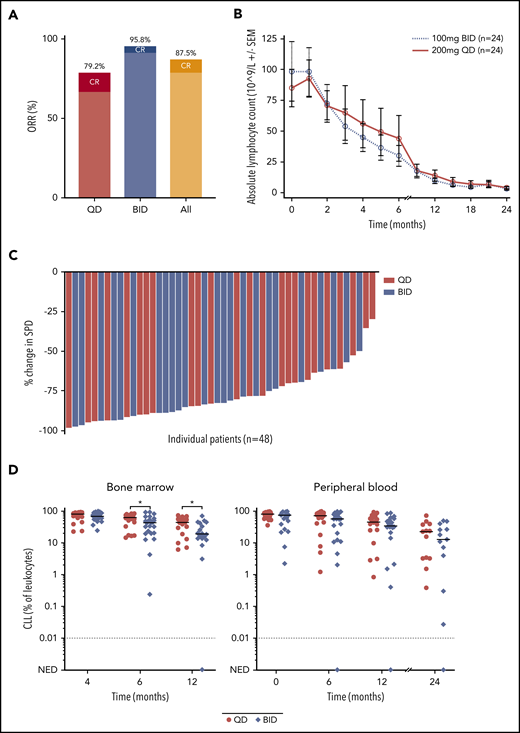
The estimated PFS rate at 24 months was 91.5% (95% CI, 70.0-97.8) in the BID group and 87.2% (95% CI, 57.2-96.7) in the QD group (Figure 3A). The estimated PFS rate at 24 months was 100% among TN patients and 84.3% (95% CI, 62.5-94.0) among R/R patients (Figure 3B). Richter transformation to Hodgkin lymphoma was detected in 2 patients and to diffuse large B-cell lymphoma in 1 patient. In the first case of Hodgkin lymphoma diagnosed at 6 months, immunohistochemical staining for CD15 and CD30 on archival bone marrow biopsy demonstrated Reed-Sternberg cells before treatment initiation.22 In the second case of Hodgkin lymphoma diagnosed at 27 months, a PLCG2 A708V mutation was detected in blood at low frequency at the time of transformation. Transformation to diffuse large B-cell lymphoma was diagnosed in 1 patient at 6 months. One patient relapsed with CLL at 24 months; a PLCG2 I671V mutation was identified at the time of progressive disease. NOTCH1 mutation was detected before treatment initiation in 3 of 4 patients who developed Richter transformation or CLL relapse. Targeted sequencing of BTK was performed prospectively in all patients at baseline, after 6 cycles, and annually on treatment. No BTK mutations have been identified.
PFS. (A) PFS in the BID group and the QD group. (B) PFS in TN patients and R/R patients.
PFS. (A) PFS in the BID group and the QD group. (B) PFS in TN patients and R/R patients.
Safety
Most treatment-emergent adverse events (TEAEs) were grade 1-2 (52.1%). The most common TEAEs were headache (68.8%), contusion (56.3%), diarrhea (43.8%), upper respiratory tract infection (41.7%), influenza-like illness (37.5%), myalgia (37.5%), arthralgia (33.3), and maculopapular rash (33.3%). TEAEs observed in ≥15% of patients are listed in Table 3. Grade ≥3 TEAEs and serious TEAEs of any grade occurred in 21 (43.8%) patients and 20 (41.7%) patients, respectively. Grade ≥3 TEAEs observed in >1 patient were neutropenia (12.5%), febrile neutropenia (4.2%), thrombocytopenia (6.3%), lung infection (8.3%), diarrhea (6.3%), and syncope (4.2%). Syncope events were attributed to diuretic use and orthostasis in 1 patient each. One patient experienced grade 5 liver failure caused by hepatitis B reactivation on day 310, which was considered related to acalabrutinib. One patient with no prior anti-CLL therapy was diagnosed with myelodysplastic syndrome (MDS) on day 156, considered unrelated to acalabrutinib, and died of refractory MDS approximately 6 months later.
TEAEs occurring in >15% of patients
| Total (n = 48) (%) | ||||||
|---|---|---|---|---|---|---|
| All grades | Grade 1 | Grade 2 | Grade 3 | Grade 4 | Grade 5 | |
| Subjects with at least 1 TEAE | 48 (100.0) | 6 (12.5) | 19 (39.6) | 17 (35.4) | 4 (8.3) | 2 (4.2) |
| Headache | 33 (68.8) | 25 (52.1) | 8 (16.7) | 0 | 0 | 0 |
| Contusion | 27 (56.3) | 27 (56.3) | 0 | 0 | 0 | 0 |
| Diarrhea | 21 (43.8) | 20 (41.7) | 1 (2.1) | 0 | 0 | 0 |
| Upper respiratory tract infection | 20 (41.7) | 2 (4.2) | 18 (37.5) | 0 | 0 | 0 |
| Influenza-like illness | 18 (37.5) | 15 (31.3) | 3 (6.3) | 0 | 0 | 0 |
| Myalgia | 18 (37.5) | 16 (33.3) | 2 (4.2) | 0 | 0 | 0 |
| Arthralgia | 16 (33.3) | 9 (18.8) | 7 (14.6) | 0 | 0 | 0 |
| Maculopapular rash | 16 (33.3) | 15 (31.3) | 0 | 1 (2.1) | 0 | 0 |
| Petechiae | 14 (29.2) | 14 (29.2) | 0 | 0 | 0 | 0 |
| Nausea | 13 (27.1) | 13 (27.1) | 0 | 0 | 0 | 0 |
| Cough | 11 (22.9) | 11 (22.9) | 0 | 0 | 0 | 0 |
| Dizziness | 11 (22.9) | 11 (22.9) | 0 | 0 | 0 | 0 |
| Nasal congestion | 11 (22.9) | 9 (18.8) | 2 (4.2) | 0 | 0 | 0 |
| Dyspnea | 10 (20.8) | 9 (18.8) | 1 (2.1) | 0 | 0 | 0 |
| Fatigue | 10 (20.8) | 9 (18.8) | 1 (2.1) | 0 | 0 | 0 |
| Pyrexia | 10 (20.8) | 9 (18.8) | 1 (2.1) | 0 | 0 | 0 |
| Neutropenia* | 9 (18.8) | 1 (2.1) | 2 (4.2) | 3 (6.3) | 3 (6.3) | 0 |
| Peripheral edema | 9 (18.8) | 9 (18.8) | 0 | 0 | 0 | 0 |
| Thrombocytopenia | 9 (18.8) | 3 (6.3) | 3 (6.3) | 2 (4.2) | 1 (2.1) | 0 |
| Lung infection | 8 (16.7) | 0 | 4 (8.3) | 4 (8.3) | 0 | 0 |
| Sinusitis | 8 (16.7) | 0 | 8 (16.7) | 0 | 0 | 0 |
| Urinary tract infection | 8 (16.7) | 0 | 7 (14.6) | 1 (2.1) | 0 | 0 |
| Total (n = 48) (%) | ||||||
|---|---|---|---|---|---|---|
| All grades | Grade 1 | Grade 2 | Grade 3 | Grade 4 | Grade 5 | |
| Subjects with at least 1 TEAE | 48 (100.0) | 6 (12.5) | 19 (39.6) | 17 (35.4) | 4 (8.3) | 2 (4.2) |
| Headache | 33 (68.8) | 25 (52.1) | 8 (16.7) | 0 | 0 | 0 |
| Contusion | 27 (56.3) | 27 (56.3) | 0 | 0 | 0 | 0 |
| Diarrhea | 21 (43.8) | 20 (41.7) | 1 (2.1) | 0 | 0 | 0 |
| Upper respiratory tract infection | 20 (41.7) | 2 (4.2) | 18 (37.5) | 0 | 0 | 0 |
| Influenza-like illness | 18 (37.5) | 15 (31.3) | 3 (6.3) | 0 | 0 | 0 |
| Myalgia | 18 (37.5) | 16 (33.3) | 2 (4.2) | 0 | 0 | 0 |
| Arthralgia | 16 (33.3) | 9 (18.8) | 7 (14.6) | 0 | 0 | 0 |
| Maculopapular rash | 16 (33.3) | 15 (31.3) | 0 | 1 (2.1) | 0 | 0 |
| Petechiae | 14 (29.2) | 14 (29.2) | 0 | 0 | 0 | 0 |
| Nausea | 13 (27.1) | 13 (27.1) | 0 | 0 | 0 | 0 |
| Cough | 11 (22.9) | 11 (22.9) | 0 | 0 | 0 | 0 |
| Dizziness | 11 (22.9) | 11 (22.9) | 0 | 0 | 0 | 0 |
| Nasal congestion | 11 (22.9) | 9 (18.8) | 2 (4.2) | 0 | 0 | 0 |
| Dyspnea | 10 (20.8) | 9 (18.8) | 1 (2.1) | 0 | 0 | 0 |
| Fatigue | 10 (20.8) | 9 (18.8) | 1 (2.1) | 0 | 0 | 0 |
| Pyrexia | 10 (20.8) | 9 (18.8) | 1 (2.1) | 0 | 0 | 0 |
| Neutropenia* | 9 (18.8) | 1 (2.1) | 2 (4.2) | 3 (6.3) | 3 (6.3) | 0 |
| Peripheral edema | 9 (18.8) | 9 (18.8) | 0 | 0 | 0 | 0 |
| Thrombocytopenia | 9 (18.8) | 3 (6.3) | 3 (6.3) | 2 (4.2) | 1 (2.1) | 0 |
| Lung infection | 8 (16.7) | 0 | 4 (8.3) | 4 (8.3) | 0 | 0 |
| Sinusitis | 8 (16.7) | 0 | 8 (16.7) | 0 | 0 | 0 |
| Urinary tract infection | 8 (16.7) | 0 | 7 (14.6) | 1 (2.1) | 0 | 0 |
Included neutropenia and neutrophil count decrease.
Seventeen patients withheld ≥7 consecutive days of acalabrutinib during the study, most often for invasive procedures or surgery (31.3%), and less commonly for adverse events (14.6%). Dose reduction for ≥3 consecutive days was required in 1 patient for grade 3 pain in multiple joints and muscles. TEAEs resulted in permanent discontinuation of acalabrutinib in 6 patients, including 3 for second primary malignancies (lung adenocarcinoma, metastatic prostate cancer, and MDS) and 1 each for autoimmune hemolytic anemia, pulmonary aspergillosis, and hepatocellular injury.
Events of clinical interest
Infections were the most frequent event of clinical interest, occurring in 39 (81.3%) patients. The most common infections were upper respiratory tract infections (41.7%), lung infection, sinusitis, and urinary tract infection (16.7% each). Eight (16.7%) patients experienced grade ≥3 infections. Viral reactivation occurred in 6 patients, 4 with herpes zoster, 1 with oral herpes simplex, and 1 with hepatitis B resulting in death. This patient was positive for anti-hepatitis B core and surface (HBs) antibodies and negative for HBs antigen and hepatitis B DNA before treatment initiation. On day 310, the patient presented with hepatocellular injury, positive HBs antigen, hepatitis B DNA of 134 million copies per milliliter, and seroconversion from positive to negative anti-HBs antibody consistent with hepatitis B reactivation. Despite treatment with entecavir, the patient died of hepatic failure on day 316. In a second patient positive for anti-hepatitis B core antibody, hepatitis B DNA, monitored every 3 months, has been undetectable to date. One patient was diagnosed with grade 2 Pneumocystis pneumonia and received treatment with oral trimethoprim-sulfamethoxazole without sequelae. One patient developed pulmonary aspergillosis in the setting of neutropenia and recent corticosteroid administration.
Our study enrolled 10 patients with history of cardiovascular disease, including atrial fibrillation, ventricular and supraventricular extrasystoles, mitral valvulopathy, and congestive cardiomyopathy. Only 1 patient experienced atrial flutter during treatment and underwent nonurgent direct current cardioversion. There has been no grade ≥3 hypertension.
Other events of clinical interest included second primary malignancies, excluding nonmelanoma skin cancer, in 6 patients and major hemorrhage from hematoma in 1 patient (supplemental Table 4).
BTK occupancy
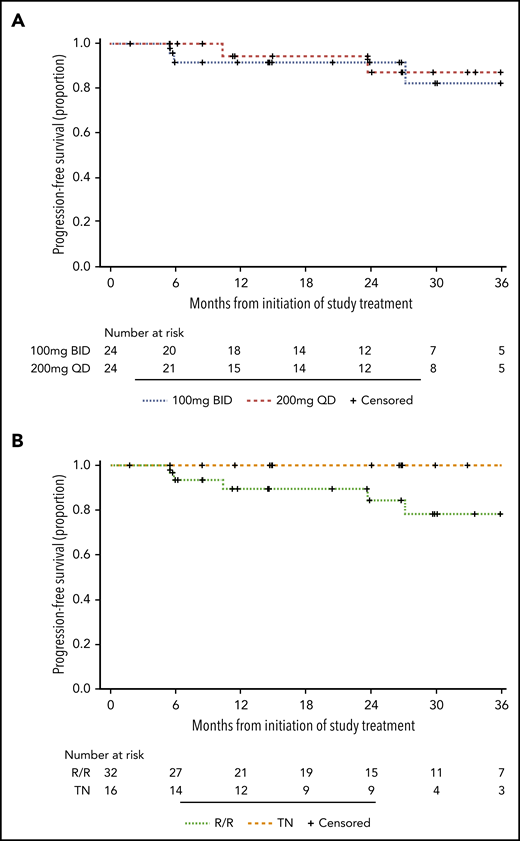
Owing to the irreversible mechanism of action of acalabrutinib, free BTK reflects the resynthesis of new BTK molecules since the last dose. To investigate the rate of BTK resynthesis, acalabrutinib was withheld for 36 to 48 hours and free BTK was measured in peripheral blood mononuclear cells collected during this period. Based on linear regression, the median estimated rate of BTK resynthesis was 14.5% per day in patients with CLL compared with 6.2% in healthy volunteers (Figure 4A).8 BTK occupancy, which is the percent of total BTK bound by acalabrutinib, was determined at drug trough (12 hours in the BID group or 24 hours in the QD group) during the first week and after 1, 6, and 12 cycles of acalabrutinib. The BID group had significantly higher BTK occupancy than the QD group at all time points (P < .05), although this difference decreased with extended treatment (Figure 4B). Given the difference in BTK occupancy between the 2 dosing groups and the administration of 100 mg BID in other acalabrutinib clinical trials, the study was amended in May 2018 to allow patients in the QD group to switch to 100 mg BID after the first 6 cycles. Seven patients elected to switch to BID dosing. BTK occupancy at drug trough increased in 5 of 6 patients tested before and after the switch in dosing schedule (Figure 4C).
BTK resynthesis rate and occupancy in PBMCs. (A) BTK resynthesis in patients with CLL (n = 48) and healthy volunteers (n = 4), shown as mean ± standard error of the mean (SEM). Linear regression was used to calculate slopes for the return of free BTK. Heathy volunteers received a 100-mg dose.8 (B) Individual BTK occupancy in the BID group and the QD group over the first 12 months of treatment. The median values are represented by the horizontal lines. *P < .05, **P < .01, ****P < .0001. (C) Individual BTK occupancy in patients before and a median of 3 months (range, 0.5-6) after switching from 200 mg QD to 100 mg BID.
BTK resynthesis rate and occupancy in PBMCs. (A) BTK resynthesis in patients with CLL (n = 48) and healthy volunteers (n = 4), shown as mean ± standard error of the mean (SEM). Linear regression was used to calculate slopes for the return of free BTK. Heathy volunteers received a 100-mg dose.8 (B) Individual BTK occupancy in the BID group and the QD group over the first 12 months of treatment. The median values are represented by the horizontal lines. *P < .05, **P < .01, ****P < .0001. (C) Individual BTK occupancy in patients before and a median of 3 months (range, 0.5-6) after switching from 200 mg QD to 100 mg BID.
Given the higher proportion of patients with TP53 aberration in the QD group compared with the BID group, we investigated the rate of BTK resynthesis and BTK occupancy based on TP53 status. There was no difference in the rate of BTK resynthesis between patients with intact TP53 (14.2% per day) and aberrant TP53 (14.8% per day; supplemental Figure 1A). Similarly, BTK occupancy at drug trough were comparable between these 2 groups of patients during the first 12 months of treatment (supplemental Figure 1B).
Little is known about the activity of acalabrutinib and other BTK inhibitors in protective niches for CLL cells.23 Therefore, we measured BTK occupancy in lymph node and bone marrow at baseline and during the first week of treatment. At drug trough, median BTK occupancy in lymph node was 95.8% (standard dose [SD], 2.9%) in the BID group and 90.1% (SD, 2.7%) in the QD group (Figure 5A). Median BTK occupancy in lymph node biopsies obtained 36 hours after dose administration fell to 85.3% (SD, 6.5%). The estimated rate of BTK resynthesis in the lymph node was 11.2% per day (Figure 5B). In paired samples from marrow and blood, BTK occupancy was measured at 4 hours and 12 hours after dosing and was not significantly different between marrow and blood. BTK occupancy in lymph node and marrow were correlated with paired blood samples (lymph node: r2 = 0.47, P < .0001, Figure 5C; marrow: r2 = 0.68, P < .0001, Figure 5D). These data indicate that acalabrutinib effectively penetrates and inhibits BTK in lymph node and marrow and that BTK resynthesis rates are comparable between blood and lymph node.
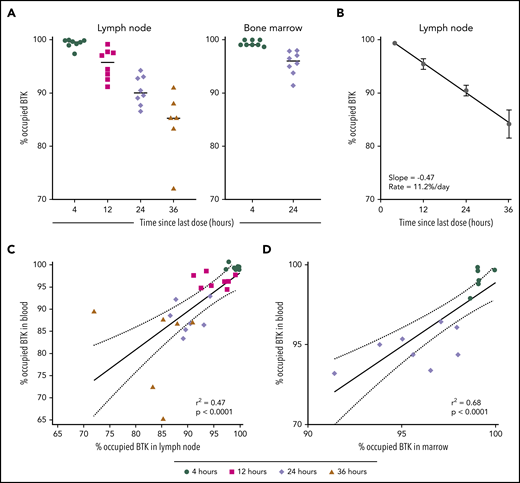
BTK occupancy in lymph node and marrow. (A) Individual BTK occupancy in the lymph node and marrow. The median values are represented by the horizontal lines. (B) BTK occupancy over time in lymph node during the acalabrutinib withholding period, shown as mean ± SEM. Linear regression was used to calculate slopes for the return of free BTK. (C-D) Correlation of BTK occupancy in paired samples of PBMC and lymph node and marrow. Shapes represent collection time since the last dose of acalabrutinib: 4 hours (●), 12 hours (▪), 24 hours (♦), and 36 hours (▲).
BTK occupancy in lymph node and marrow. (A) Individual BTK occupancy in the lymph node and marrow. The median values are represented by the horizontal lines. (B) BTK occupancy over time in lymph node during the acalabrutinib withholding period, shown as mean ± SEM. Linear regression was used to calculate slopes for the return of free BTK. (C-D) Correlation of BTK occupancy in paired samples of PBMC and lymph node and marrow. Shapes represent collection time since the last dose of acalabrutinib: 4 hours (●), 12 hours (▪), 24 hours (♦), and 36 hours (▲).
Transcriptome analysis
To understand the biological consequences of BTK inhibition, RNA-seq was performed on purified tumor cells from blood at baseline and after 1 cycle and 6 cycles of treatment in 20 patients. Samples were separated by time on treatment on principal component analysis (Figure 6A). Compared with baseline, 148 genes were differentially expressed (DE; fold-change ≥2, false discovery rate [FDR] <0.1) after 1 cycle and 360 genes after 6 cycles (supplemental Table 5). Approximately 75% of DE genes after 1 cycle overlapped with DE genes after 6 cycles. In addition, a progressive change in the expression of DE genes was observed during the first 6 cycles (Figure 6B). Pathway enrichment analysis showed downregulation of BCR and nuclear factor-κB pathways, cytokine signaling, and cellular metabolism with acalabrutinib therapy (FDR < 0.05, Figure 6C; supplemental Table 6). No pathways were found to be upregulated on treatment.
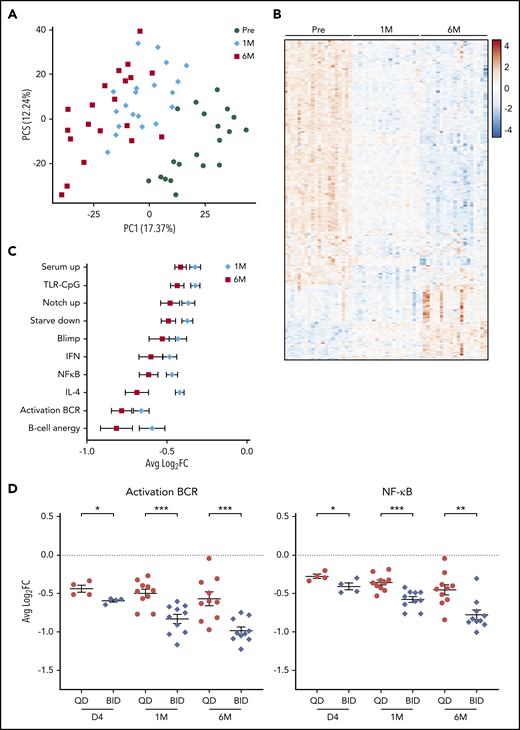
Transcriptome analysis of circulating tumor cells from patients before and after treatment with acalabrutinib. (A) Principal component analysis of samples collected at baseline (●), after 1 cycle (♦), and after 6 cycles (▪). (B) Heatmap of DE genes (fold-change ≥2, FDR < 0.1) (C) Average ± SEM log2 fold-change in expression of leading-edge genes of the indicated gene signatures after 1 cycle and after 6 cycles compared with baseline. (D) Average ± SEM log2 fold-change in leading edge genes from BCR and nuclear factor-κB signatures in the BID group compared with the QD group. *P < .05, **P < .01, ***P < .001.
Transcriptome analysis of circulating tumor cells from patients before and after treatment with acalabrutinib. (A) Principal component analysis of samples collected at baseline (●), after 1 cycle (♦), and after 6 cycles (▪). (B) Heatmap of DE genes (fold-change ≥2, FDR < 0.1) (C) Average ± SEM log2 fold-change in expression of leading-edge genes of the indicated gene signatures after 1 cycle and after 6 cycles compared with baseline. (D) Average ± SEM log2 fold-change in leading edge genes from BCR and nuclear factor-κB signatures in the BID group compared with the QD group. *P < .05, **P < .01, ***P < .001.
Lymph node samples obtained at baseline and during the first week of treatment were also subjected to RNA-seq. Similar to circulating tumor cells, multiple oncogenic pathways were prominently downregulated in the lymph node of patients treated with acalabrutinib (supplemental Table 7). Additionally, gene signatures specific to the tumor microenvironment, such as proliferation and T-cell activation, were suppressed in on-treatment samples.
Given the difference in BTK occupancy between the BID and QD groups, we compared transcriptome data between the dosing groups. Multiple pathways were more potently downregulated after 3 days of acalabrutinib in the BID group than in the QD group (Figure 6D; supplemental Figure 2). With extended treatment, the difference in the inhibition of most pathways between the BID group and the QD group became more pronounced (Figure 6D; supplemental Figure 2). The degree of BCR pathway inhibition was significantly correlated with BTK occupancy (BCR: r2 = 0.53, P = .04; supplemental Figure 3).
Discussion
This phase 2 study was designed to comprehensively assess the pharmacodynamic effects of acalabrutinib over time and across tissue compartments. Key findings include the rate of BTK resynthesis, superior target inhibition with more frequent dosing, and the reinforcement of biological effects related to minor but persistent differences in BTK occupancy.
In untreated CLL, BTK mRNA and protein expression is higher than in normal B cells.24 We report, for the first time, the rate of BTK resynthesis in patients with CLL. Following irreversible inhibition by acalabrutinib, BTK was resynthesized at a faster rate in CLL cells compared with normal B cells. The improvement in BTK occupancy over the first year of treatment suggests that continuous BTK inhibition decelerates BTK resynthesis. which could reflect the suppression of global mRNA translation induced by BCR signaling as shown by prior in vitro studies.10 The difference in BTK occupancy between the BID group and the QD group diminished, but remained significant (P < .05), with extended treatment. BTK occupancy improved in most patients who switched from QD to BID administration, even after more than 6 cycles of treatment.
CLL cells depend on interactions with their microenvironment for survival and proliferation. Cell culture conditions that simulate the tumor microenvironment protect CLL cells from spontaneous and drug-induced apoptosis.25 Because of ease of access, most pharmacodynamic analyses, including that of BTK inhibitor therapy, have been limited to blood samples. In our study, high BTK occupancy was achieved in lymph node and marrow, demonstrating the activity of acalabrutinib in tumor protective niches. Simultaneous measurements from blood and lymph node showed that BTK occupancy in blood approximates, and could be used to infer, BTK occupancy in lymph node.
The degree of BTK inhibition required to suppress survival and growth signals in CLL cells is unknown. Acalabrutinib therapy resulted in progressive downregulation of pathogenic pathways, consistent with our previous RNA-seq analysis of patients treated with ibrutinib.21 A comparison of the BID group and the QD group revealed a striking difference between BTK occupancy and the downstream effects of BTK inhibition. Multiple pathways were more potently downregulated in the BID group than in the QD group. Inhibition of these pathways strengthened over the first 6 cycles of treatment and to a greater degree in the BID group. Thus, stable and near-complete BTK occupancy with BID dosing reinforces the suppression of oncogenic pathways in CLL cells.
Acalabrutinib yielded a high ORR in patients with CLL across prognostic subgroups. Responses deepened with extended treatment, but complete remissions were only achieved by 4 patients (8.3%). After a median of 25.5 months on treatment, 89.6% of patients remain alive and progression-free. These efficacy results are consistent with the phase 1-2 study of acalabrutinib for relapsed CLL.1 As one of the earliest clinical investigations of acalabrutinib, this study aimed to assess the overall activity and pharmacodynamics of acalabrutinib, but was not powered to detect a difference in clinical outcomes between BID and QD dosing. Notwithstanding, flow cytometry of bone marrow after 6 and 12 months of treatment showed less disease burden in the BID group than in the QD group. Four patients developed disease progression during treatment with acalabrutinib, 1 in the QD group and 3 in the BID group. In 2 patients, we identified mutations in PLCG2 (I671V and A708V) located in the autoinhibitory SH2 domain, a known hotspot for mutations associated with ibrutinib resistance. Disruption of the SH2 domain in PLCG2 results in a gain of function and reactivation of BCR signaling,26,27 causing CLL relapse. Neither mutation has been previously reported, consistent with the heterogenous nature of PLCG2 mutations on BTK inhibitor therapy.
Fewer toxicities related to off-target kinase inhibition are expected because of the increased kinase selectivity of acalabrutinib. In contrast to ibrutinib, which is associated with an incidence of atrial fibrillation or flutter of 11.2% in randomized controlled trials,28 only 1 patient in this current study developed atrial flutter during treatment with acalabrutinib. The seemingly lower incidence of arrhythmia could be an important consideration in the selection of a BTK inhibitor, especially for patients with underlying cardiovascular disease. Conversely, nearly 70% of patients experienced grade 1-2 headache, most often during the first cycles and resolved with extended treatment. Other adverse events observed on this study, such as bleeding diathesis, diarrhea, myalgia, and infections, were similar to the reported safety profile of ibrutinib. Given the different kinase selectivity of acalabrutinib and ibrutinib, some overlapping toxicities could, at least in part, be related to the inhibition of BTK rather than off-target kinases.
In summary, acalabrutinib is an effective and safe treatment of relapsed or refractory or high-risk treatment naïve CLL. Twice daily dosing increases BTK occupancy and downregulation of oncogenic pathways. Sustained and complete target occupancy is required for maximal inhibition of tumor cells although an absolute threshold for complete occupancy remains ill-defined. Extended follow-up is required to determine its potential effect on long-term clinical outcomes.
Presented, in part, in abstract form at the 60th annual meeting of the American Society of Hematology, San Diego, CA, 1-4 December 2018.
Requests for deidentified patient data can be submitted to Acerta Pharma 3 months after publication for a period of 5 years after the publication date. Full data disclosure is provided in supplemental Methods. RNA-seq data are available in the Gene Expression Omnibus data repository (accession number GSE136634).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the patients, their families, all associate investigators, site personnel, the Collaborative Health Initiative Research Program at Uniformed Services University of the Health Sciences, National Heart, Lung, and Blood Institute (NHLBI) Sequencing and Genomics Core, NHLBI Bioinformatics and Computational Biology Core, National Institutes of Health Interventional Radiology Section, and Gary De Jesus from Acerta Pharma for statistical analysis of Bruton tyrosine kinase occupancy.
This study was sponsored by Acerta Pharma, a member of the AstraZeneca group and supported, in part, by the Intramural Research Program of the National Institutes of Health National Heart, Lung, and Blood Institute. A.W. received research funding from Acerta Pharma, BV.
Authorship
Contribution: Conceptualization performed by S.E.M.H., M.Z.H.F., R.I., A.H., and A.W.; formal analysis done by C.S., E.K.K., J.C., M.G., and M.H.W.; investigation undertaken by C.S., P.N., J.C., M.G., C.P., I.E.A., M.S.-S., C.M.Y., I.M., H.M.H., S.P., and A.W.; data curation by C.S. and P.N.; writing of the original draft performed by C.S. and A.W.; writing review and editing performed by all authors; visualization performed by C.S., E.K.K., J.C., and M.G.; supervision undertaken by C.S., P.P., M.Z.H.F., T.C., and A.W.; project administration done by P.N. and E.M.G.; and funding acquisition performed by A.W.
Conflict-of-interest disclosure: J.C., M.G., A.H., R.I., M.H.W., P.P., and T.C. were or currently are full-time employees and shareholders of Acerta Pharma, BV. M.Z.H.F. is currently a full-time employee of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, and shareholder of Merck & Co., Inc., Kenilworth, NJ. The remaining authors declare no competing financial interests.
Correspondence: Adrian Wiestner, Hematology Branch, NHLBI, NIH, Bldg 10, CRC 3-5140, 10 Center Dr, Bethesda, MD 20892-1202; e-mail: wiestnea@nhlbi.nih.gov.
