

Key Points
A new series of ceramide analogs were synthesized, identifying some that effectively inhibit PEL growth and progression in vitro and in vivo.
The mechanisms of new ceramide analogs, including novel sphingolipid-regulated cellular genes required for PEL survival, were identified.

Abstract
Primary effusion lymphoma (PEL) is an aggressive malignancy with poor prognosis even under chemotherapy. Kaposi sarcoma–associated herpesvirus (KSHV), one of the human oncogenic viruses, is the principal causative agent. Currently, there is no specific treatment for PEL; therefore, developing new therapies is of great importance. Sphingolipid metabolism plays an important role in determining the fate of tumor cells. Our previous studies have demonstrated that there is a correlation between sphingolipid metabolism and KSHV+ tumor cell survival. To further develop sphingolipid metabolism-targeted therapy, after screening a series of newly synthesized ceramide analogs, here, we have identified compounds with effective anti-PEL activity. These compounds induce significant PEL apoptosis, cell-cycle arrest, and intracellular ceramide production through regulation of ceramide synthesizing or ceramide metabolizing enzymes and dramatically suppress tumor progression without visible toxicity in vivo. These new compounds also increase viral lytic gene expression in PEL cells. Our comparative transcriptomic analysis revealed their mechanisms of action for inducing PEL cell death and identified a subset of novel cellular genes, including AURKA and CDCA3, controlled by sphingolipid metabolism, and required for PEL survival with functional validation. These data provide the framework for the development of promising sphingolipid-based therapies against this virus-associated malignancy.
Introduction
Kaposi sarcoma–associated herpesvirus (KSHV) is a principal causative agent of several cancers arising in AIDS and/or other immunocompromised patients.1 One of these cancers, primary effusion lymphoma (PEL), harbors KSHV episome in transformed B cells and typically presents as effusions in the pleural, pericardial, or abdominal cavities of patients, although solid/extracavitary lesions have also been reported.2,3 PEL cells are pleomorphic with features of large immunoblastic, plasmablastic, or anaplastic cells.3,4 All cases of PEL have KSHV infection of tumor cells, and 80% have Epstein-Barr virus (EBV) coinfection.5 Combination chemotherapies, such as EPOCH (modified infusional etoposide, vincristine, and doxorubicin with cyclophosphamide and prednisone), represent the current standard for PEL treatment.6-8 Recent studies suggest that some immunomodulatory drugs, such as lenalidomide and pomalidomide, may present a promising treatment strategy against PEL.9,10 A recent study reported that the current median overall survival length in treated PEL patients is 22 months.11 They also found that inflammatory cytokines (eg, interleukin-6) and tumor EBV status are the strongest prognostic factors for PEL patients. Therefore, there is still an urgent need to identify novel targets and develop more effective treatment strategies against PEL progression.
The initial step in the synthesis of bioactive sphingolipids is the hydrolysis of ceramide to form sphingosine. In cells, sphingosine can be phosphorylated by sphingosine kinases (SphK1 or SphK2) to form sphingosine-1-phosphate (S1P).12 The relative ratio of ceramide to S1P is an important determinant of cell survival, with higher ratios promoting apoptosis and lower ratios inducing proliferation.12,13 Thus, ceramide is referred to as a “tumor suppressor lipid,” since it powerfully potentiates signaling events that drive apoptosis, cell-cycle arrest, and autophagic response.12,14 Because of their pleiotropic roles, targeting bioactive sphingolipids has evolved as a promising therapeutic approach for cancer treatment over the past 2 decades.15 One common approach to promote apoptosis in cancer cells is the use of exogenous ceramide analogs or mimetics as therapeutic agents. Another effective method is increasing intracellular ceramide concentrations to regulate cancer cell growth. Certain chemotherapeutic agents, such as daunorubicin, camptothecin, fludarabine, etoposide, and gemcitabine, increase ceramide generation through the de novo ceramide pathway or via activation of SMases.12 In addition, some small molecule inhibitors of important metabolic enzymes involved in the sphingolipid pathway have been used to induce the accumulation of ceramide in cancer cells in vitro and in vivo.15,16
We have recently established that sphingolipid metabolism, especially intracellular balance of ceramide/S1P, plays an important role in PEL survival.17-19 For example, we demonstrated that ABC294640, the only first-in-class orally available inhibitor of SphK2, selectively induces caspase-dependent apoptosis and cell death of KSHV+ PEL cell lines.17 Interestingly, we found that KSHV infection increased SphK2 expression from host cells, which may render KSHV-infected cells more sensitive to SphK2 inhibitor.20 Subsequently, we determined that some exogenous short-/long-chain ceramides, such as C2-, C8-, and C18-Cer, also induce the apoptosis for PEL cell lines.18 To further develop sphingolipid metabolism-targeted therapies, we have designed and synthesized a series of new ceramide analogs by introducing sulfonamide, amide, and/or certain aromatic systems in the ceramide side chain or backbone. These chemical modifications potentially increase the antiproliferation activity, the regulation of ceramide metabolism, and the stability of the ceramide analogs. In the current study, we screened these new ceramide analogs and identified the ones displaying effective anti-PEL activity in vitro and in vivo through inducing tumor cell apoptosis and cell-cycle arrest. We identified the mechanisms involved in these selective ceramide analogs, increasing intracellular ceramide levels through a variety of ceramide synthases or ceramide metabolism enzymes. Our transcriptomic analysis revealed cellular gene profiles altered by new ceramide analogs treatment and identified a subset of cellular genes controlled by sphingolipid metabolism and required for PEL cell survival. Thus, our study has demonstrated a promising strategy for the potential development of sphingolipid metabolism-targeted therapy against this virus-associated malignancy.
Materials and methods
Cell culture and reagents
KSHV+ PEL cell line, BCBL-1, was kindly provided by Dean Kedes (University of Virginia) and cultured as described previously.17 All the other PEL cell lines, BCP-1, BC-1, BC-3, and JSC-1, multiple myeloma cell lines, RPMI-8266, and U266 were purchased from American Type Culture Collection and cultured as recommended by the manufacturer. The diffuse large B-cell lymphoma cell lines, Su-DHL-6 and HBL-1, were kindly gifted by Margot Thome at University of Lausanne. Human peripheral blood mononuclear cells were isolated from whole blood from 2 healthy donors following Ficoll gradient separation. Isolation of circulating human B cells from peripheral blood mononuclear cells was performed as described previously.17 For both donors, 92% to 95% pure populations of CD19+ cells were recovered (data not shown). All the cells were cultured at the conditions of 37°C with 5% CO2. ABC294640 was purchased from SelleckChem. All new ceramide analogs were synthesized by Maryam Foroozesh’s group at Xavier University of Louisiana.
New ceramide analogs 315 and 403 synthesis
For the synthesis of analog 315, Boc-serine 1 (N-(tert-butoxycarbonyl)-d-serine) was coupled with tetradecylamine 2 to get the corresponding amide 3. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide and hydroxybenzotriazole were used as coupling reagents, and N-methyl-morpholine was used as a base. Tetrahydrofuran was used as a solvent. The Boc group was then removed using trifluoracetic acid to give the free amine 4, which was further reacted with salicylaldehyde in the presence of the strong base sodium hydroxide in methanol (solvent) to get analog 315.21,22 For the synthesis of analog 403, d-sphingosine was reacted with phenylacetyl chloride in the presence of sodium bicarbonate as a base and dichloromethane as solvent to get analog 403.23
Cell proliferation and apoptosis assays
Cell proliferation was measured using the WST-1 Assay (Roche, Indianapolis, IN). Briefly, after the period of treatment of cells, 10 μL per well of cell proliferation reagent, WST-1 (4-[3-(4-iodophenyl)-2-(4-nitro-phenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate) was added to 96-well microplates and incubated for 3 hours at 37°C in 5% CO2. The absorbance of samples was measured by using a microplate reader at 490 nm. Flow cytometry was used for the quantitative assessment of apoptosis with the fluorescein isothiocyanate–Annexin V/propidium iodide (PI) Apoptosis Detection Kit I (BD Pharmingen, San Jose, CA) and analyzed on a FACSCalibur 4-color flow cytometer (BD Bioscience, San Jose, CA).
Cell-cycle analysis
PEL cell pellets were fixed in 70% ethanol and incubated at 4°C overnight. Cell pellets were resuspended in 0.5 mL of 0.05 mg/mL PI plus 0.2 mg/mL RNaseA and incubated at 37°C for 30 minutes. Cell-cycle distribution was analyzed using a flow cytometer as mentioned above.
Immunoblotting
Total cell lysates (20 μg) were resolved by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes, and immunoblotted with antibodies for cleaved Caspase3/9, Bax, XIAP, Cyclin D1, CDK6, phosphor-Rb, p21, SphK1, SphK2, GCS, AURKA, CDCA3, MYC (Cell Signaling, Danvers, MA), and β-actin (Sigma, St. Louis, MO) for loading controls. Immunoreactive bands were identified using an enhanced chemiluminescence reaction (Perkin-Elmer, Waltham, MA), visualized by autoradiography, scanned, and quantified using the ImageJ Software.
Sphingolipid analyses
Quantification of sphingolipid species was performed using a Thermo Finnigan TSQ 7000 triple-stage quadruple mass spectrometer operating in multiple-reaction monitoring positive ionization mode (Thermo Fisher Scientific, Waltham, MA). Quantification was based on calibration curves generated by spiking an artificial matrix with known amounts of target standards, and an equal amount of the internal standard. The target analyte:internal standard peak area ratios from each sample were compared with the calibration curves using linear regression. Final results were expressed as the ratio of sphingolipid normalized to total phospholipid phosphate level using the Bligh and Dyer lipid extract method.24
Safe dosing in vivo study
For drug delivery, ceramide analog 315 or 403 was diluted in sterile polyethylene glycol (95%):dimethyl sulfoxide (5%) to achieve 100 μL total volume. Six- to 8-week-old male nonobese diabetic/severe-combined immunodeficiency (NOD/SCID) mice (Jackson Laboratory, Ellsworth, ME) received intraperitoneal (IP) injections with a ceramide analog (12.5, 25.0, or 50.0 mg/kg, respectively) or vehicle, once daily, 2 days per week for 4 weeks. Weights were recorded weekly. Two experiments, with 5 mice per group for each experiment, were performed. The immediate and delayed toxicity of these treatments were assessed by careful monitoring of the animals for signs of toxicity, including respiratory difficulties, feeding or drinking difficulties, evidence of spastic paralysis, convulsion, or blindness. At the end of treatment, the different organ tissues (liver, kidney, heart, lung, and spleen) were collected from the representative control, vehicle, or ceramide analogs–treated mice. Formalin-fixed, paraffin-embedded tissues were microtome-sectioned to a thickness of 4 µm, placed on electromagnetically charged slides, and stained with hematoxylin and eosin (H&E) for routine histologic analysis.
PEL xenograft model
Aliquots of 107 BCBL-1 cells were diluted in 200 μL sterile phosphate-buffered saline, and 6- to 8-week-old male NOD/SCID mice received IP injections with a single aliquot. Ceramide analog solution or vehicle was administered initially at 24 hours after BCBL-1 injections and continued once daily, 2 days per week for 5 weeks. Two experiments, with 5 mice per group for each experiment, were performed. Weights were recorded weekly as a surrogate measure of tumor progression, and ascites fluid volumes were tabulated for individual mice at the completion of each experiment. All protocols were approved by the University of Arkansas for Medical Sciences Animal Care and Use Committee in accordance with national guidelines. Cell apoptosis in the ascites samples collected from the representative vehicle- or ceramide analog-treated mice was detected using the TUNEL Assay Kit (Abcam, Cambridge, MA).
Microarray
BCP-1 and BCBL-1 cells were treated with vehicle, 315, 403, or ABC294640 for 48 hours, respectively. Total RNA was isolated using the Qiagen RNeasy Kit (Qiagen, Germantown, MD), and 500 ng of total RNA was used to synthesize double-stranded complementary DNA. Biotin-labeled RNA was generated using the TargetAmp-Nano Labeling Kit for Illumina Expression BeadChip (Epicentre, Madison, WI), according to the manufacturer’s instructions, and hybridized to the Human HT-12 v4 Expression BeadChip (Illumina, San Diego, CA). Using Illumina’s GenomeStudio software, signals were normalized using a “cubic spline algorithm” that assumes that the distribution of transcript abundance is similar in all samples. Background signals were removed using the “detection P value algorithm” to remove targets with signal intensities equal to or lower than that of irrelevant probes (with no known targets in the human genome but thermodynamically similar to the relevant probes). The microarray experiments were performed twice for each group, and the average values were used for analysis. Common and unique sets of genes and enrichment analysis were performed using MetaCore Software (Thompson Reuters).
RNA interference (RNAi) assays
For RNAi assays, AURKA, CDCA3, SphK1, or SphK2 On-Target plus SMART pool small interfering RNA (siRNA; Dharmacon) or negative control siRNA was delivered using the DharmaFECT Transfection Reagent as recommended by the manufacturer.
Quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Total cellular RNA was isolated and purified using the RNeasy Mini Kit (QIAGEN). Complementary DNA was synthesized using SuperScript III First-Strand Synthesis SuperMix Kit (Invitrogen). Primers used for amplification of target genes are listed in supplemental Table 1, available on the Blood Web site. Amplification was performed on an iCycler IQ Real-Time PCR Detection System and analyzed as described previously.25
Statistical analysis
Significance for differences between experimental and control groups was determined using the 2-tailed Student t test, and P values <.05 or .01 were considered significant or highly significant, respectively. The 50% inhibitory concentrations (IC50) were calculated by using SPSS 20.0.
Results
Identification of new ceramide analogs with effective anti-PEL activity
Using the WST-1 Cell Proliferation Assay, we initially screened a series of new ceramide analogs (a total of 19 new compounds for which the structures are shown in Figure 1A), and identified 6 [(S,E)-3-hydroxy-2-((4-nitrobenzylidene)amino)-N-tetradecylpropanamide (compound 301), (S,E)-2-((2-chlorobenzylidene)amino)-3-hydroxy-N-tetradecylpropanamide (compound 309), (S,E)-3-hydroxy-2-((4-hydroxybenzylidene)amino)-N-tetradecylpropanamide (compound 313), (S,E)-3-hydroxy-2-((2-hydroxybenzylidene)amino)-N-tetradecylpropanamide (compound 315), N-((2S,3R,E)-1,3-dihydroxyoctadec-4-en-2-yl)-2-phenylacetamide (compound 403) and (S)-3-hydroxy-2-((4-methylphenyl)sulfonamide)-N-tetradecylpropanamide (compound 953)] that displayed the most effective anti-PEL activities (>70% inhibition rate; Figure 1B). We next selected two of them, 315 and 403, for subsequent experiments due to their high efficacy observed in the initial screening and relative stability (their synthetic pathways are shown in Figure 2A-B and described in the “Methods”). Using a range of doses, we found that both analogs showed obvious dose-dependent inhibition of growth of the PEL cell lines, BCBL-1 and BCP-1, derived from cancer patients (Figure 2C-F). In addition, both analogs displayed similar inhibitory effects on other PEL cell lines, such as BC-1, BC-3, and JSC-1 (supplemental Figure 1). Notably, the IC50 ranges of analogs 315 and 403 in PEL cell lines are ∼4 to 9 µM, which are much lower than those of other reported sphingolipid metabolism-targeted molecules, such as the exogenous short-/long-chain ceramides and the SphK2 inhibitor (ABC294640) from our previous studies17,18 (Figure 2G).
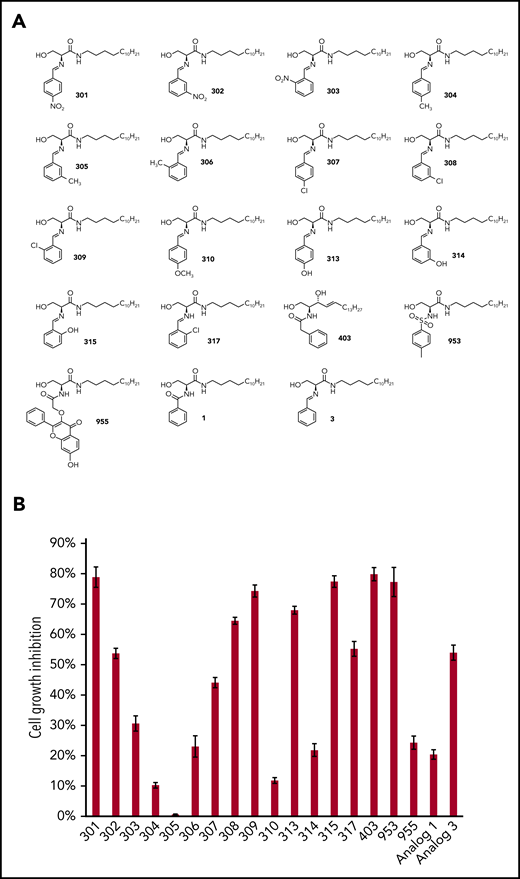
The screening of new ceramide analogs with anti-PEL activities. (A) The chemical structures of our newly synthetic ceramide analogs. (B) KSHV+ PEL cell line, BCBL-1, was treated with 20 µM of analogs or vehicle for 48 hours. The cell proliferation status was then examined using the WST-1 Cell Proliferation Assay (Roche). The cell growth inhibitory rates were calculated by comparing to the vehicle-treated controls. Error bars represent standard deviation (SD) for 3 independent experiments.
The screening of new ceramide analogs with anti-PEL activities. (A) The chemical structures of our newly synthetic ceramide analogs. (B) KSHV+ PEL cell line, BCBL-1, was treated with 20 µM of analogs or vehicle for 48 hours. The cell proliferation status was then examined using the WST-1 Cell Proliferation Assay (Roche). The cell growth inhibitory rates were calculated by comparing to the vehicle-treated controls. Error bars represent standard deviation (SD) for 3 independent experiments.
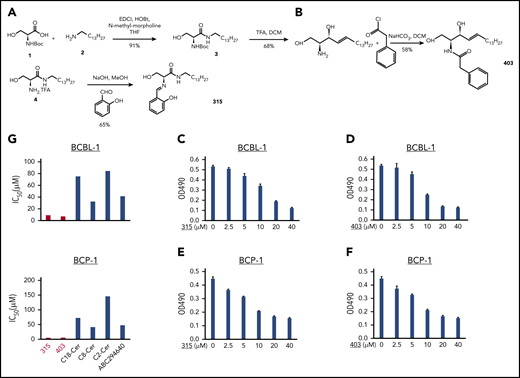
New ceramide analogs 315 and 403 effectively reduce PEL proliferation in a dose-dependent manner. (A-B) The synthetic schemes for ceramide analogs 315 and 403. (C-F) KSHV+ PEL cell lines, BCBL-1 (C-D) and BCP-1 (E-F), were treated with the indicated concentrations of analog 315, 403, or vehicle for 48 hours. The cell proliferation status was then examined using the WST-1 Cell Proliferation Assay (Roche). Error bars represent SD for 3 independent experiments. (G) Comparison of 50% IC50 of different compounds targeting sphingolipid metabolism in PEL cell lines. DCM, dichloromethane; THF, tetrahydrofuran.
New ceramide analogs 315 and 403 effectively reduce PEL proliferation in a dose-dependent manner. (A-B) The synthetic schemes for ceramide analogs 315 and 403. (C-F) KSHV+ PEL cell lines, BCBL-1 (C-D) and BCP-1 (E-F), were treated with the indicated concentrations of analog 315, 403, or vehicle for 48 hours. The cell proliferation status was then examined using the WST-1 Cell Proliferation Assay (Roche). Error bars represent SD for 3 independent experiments. (G) Comparison of 50% IC50 of different compounds targeting sphingolipid metabolism in PEL cell lines. DCM, dichloromethane; THF, tetrahydrofuran.
New ceramide analogs induce apoptosis and cell-cycle arrest for PEL cells
We next sought to determine the underlying mechanisms for anti-PEL activity of these new ceramide analogs. Using flow cytometry, we found that both analogs 315 and 403 induced apoptosis in a dose-dependent fashion for BCBL-1 and BCP-1 cells (Figure 3A-C). In contrast to the PEL cells, there was no discernible drug-induced toxicity of analogs 315 and 403 at 10 µM and 20 µM for primary human CD19+ B cells isolated from peripheral blood of healthy donors (supplemental Figure 2). We also observed the increased cleavage of caspases-3 and -9 and the expression of proapoptotic protein, Bax, from PEL cell lines exposed to these compounds (Figure 3D-E). In addition, we observed a decreased expression of X-linked inhibitor of apoptosis protein, a physiologic substrate of Akt that is stabilized to inhibit programmed cell death and has a direct effect on caspase-3 and -9.26
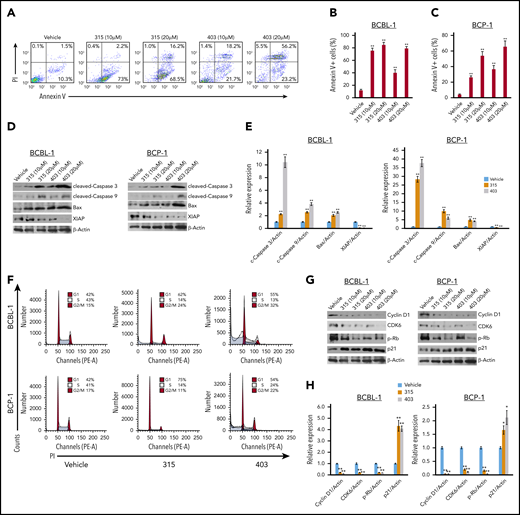
New ceramide analogs 315 and 403 induce significant PEL apoptosis and cell-cycle arrest. (A-C) BCBL-1 (A-B) and BCP-1 (C) cells were treated with the indicated concentrations of analog 315, 403, or vehicle for 24 hours. Cell viability and apoptosis were then measured by Annexin V-PI staining and flow cytometry analysis. (D-E) Protein expression was detected using immunoblots, and β-actin was used as a loading control. (F-H) BCBL-1 and BCP-1 cells were treated with 10 µM of analog 315, 403, or vehicle for 24 hours and then stained by PI and analyzed by flow cytometry. Representative results from one of 3 independent experiments are shown. Protein expression was detected as above. Error bars represent SD for 3 independent experiments. *P < .05; **P < .01.
New ceramide analogs 315 and 403 induce significant PEL apoptosis and cell-cycle arrest. (A-C) BCBL-1 (A-B) and BCP-1 (C) cells were treated with the indicated concentrations of analog 315, 403, or vehicle for 24 hours. Cell viability and apoptosis were then measured by Annexin V-PI staining and flow cytometry analysis. (D-E) Protein expression was detected using immunoblots, and β-actin was used as a loading control. (F-H) BCBL-1 and BCP-1 cells were treated with 10 µM of analog 315, 403, or vehicle for 24 hours and then stained by PI and analyzed by flow cytometry. Representative results from one of 3 independent experiments are shown. Protein expression was detected as above. Error bars represent SD for 3 independent experiments. *P < .05; **P < .01.
Next, we found that both analogs caused obvious PEL cell-cycle G1 and/or G2/M arrest when compared with the vehicle control by flow cytometry analysis (Figure 3F). Further analysis indicated that treatment with 315 and 403 affected the expression of several cell-cycle checkpoint regulatory proteins (positive or negative), such as decreasing Cyclin D1, CDK6, phosphor-Rb, while increasing p21 from PEL cell lines (Figure 3GH).
New ceramide analogs increase intracellular ceramide levels in PEL cells through regulation of ceramide synthesizing and/or metabolizing enzymes
Mass spectrometric–based lipidomics analysis was used to quantify and compare intracellular levels of bioactive ceramide species in PEL cell lines treated with or without new ceramide analogs. We found that both analogs 315 and 403 increased total levels of intracellular ceramides (2.5- to 4.0-folds) in BCBL-1 and BCP-1 cell lines when compared with the vehicle. The lipidomics analysis of individual ceramide species indicated that most of the species, including those from C14-Cer to C26-Cer, were upregulated in the PEL cell lines exposed to 315 and 403, although the extent of the increase varies among the ceramide species and cell lines (Figure 4A-D). Based on the lipidomics analyses, the most upregulated ceramide species were C16-, C18:1-, C18-, and C20-Cer in PEL cell lines treated with either analog 315 or 403. To better understand the significance of the observed changes in ceramides, we compared the composition and proportion of individual ceramide species present in PEL cell lines that were untreated or treated with the ceramide analogs. The predominant ceramide species without treatment were similar in the BCBL-1 and BCP-1 cells and included C16-, C22-, C24:1-, and C24-Cer (Figure 4E-F). Importantly, treating the PEL cells with either 315 or 403 mainly altered the proportion of C16-, C18-, C24:1-, and C24-Cer.
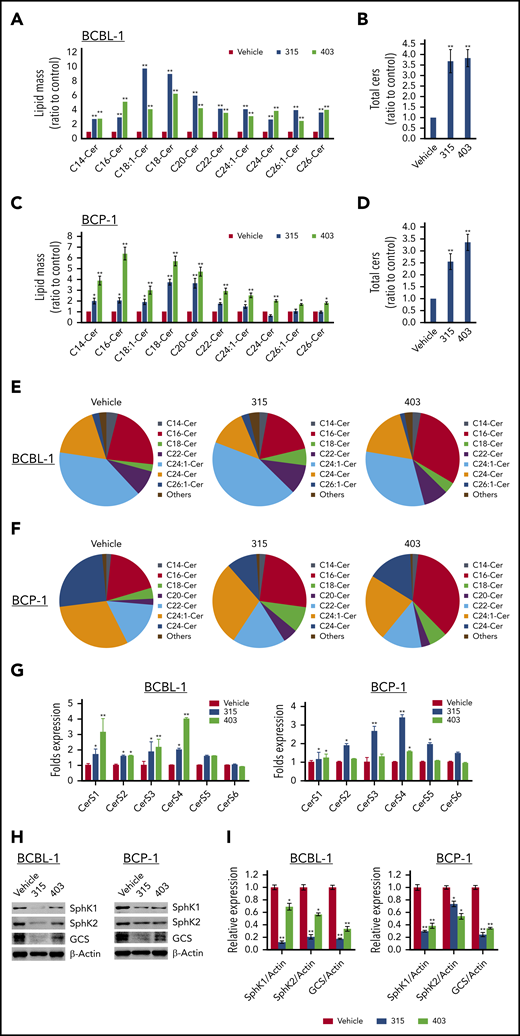
New ceramide analogs 315 and 403 induce intracellular ceramides production through regulation of ceramide synthases and enzymes of ceramide metabolism. (A-D) BCBL-1 (A-B) and BCP-1 (C-D) cells were treated with 20 µM of analog 315, 403, or vehicle for 24 hours. Ceramide species and total levels were then quantified using lipidomics analyses as described in the “Methods.” Error bars represent SD for 2 independent experiments. *P < .05; **P < .01. (E-F) Relative compositions and proportions of specific ceramide species were present within vehicle-, 315-, or 403-treated PEL cell lines. Each color representing a specific ceramide species is labeled beside the pie chart. (G) BCBL-1 and BCP-1 cells were treated with 20 µM of analog 315, 403, or vehicle for 24 hours; then the gene transcription was quantified using qRT-PCR. (H-I) The protein expression was detected using immunoblots. Error bars represent SD for 3 independent experiments. *P < .05; **P < .01.
New ceramide analogs 315 and 403 induce intracellular ceramides production through regulation of ceramide synthases and enzymes of ceramide metabolism. (A-D) BCBL-1 (A-B) and BCP-1 (C-D) cells were treated with 20 µM of analog 315, 403, or vehicle for 24 hours. Ceramide species and total levels were then quantified using lipidomics analyses as described in the “Methods.” Error bars represent SD for 2 independent experiments. *P < .05; **P < .01. (E-F) Relative compositions and proportions of specific ceramide species were present within vehicle-, 315-, or 403-treated PEL cell lines. Each color representing a specific ceramide species is labeled beside the pie chart. (G) BCBL-1 and BCP-1 cells were treated with 20 µM of analog 315, 403, or vehicle for 24 hours; then the gene transcription was quantified using qRT-PCR. (H-I) The protein expression was detected using immunoblots. Error bars represent SD for 3 independent experiments. *P < .05; **P < .01.
There are 3 primary pathways that generate intracellular ceramide: the sphingomyelinase pathway, the de novo pathway, and the salvage pathway.14,27 Ceramide synthesis by both the de novo and the salvage pathways depends on the activity of one or more Ceramide synthases (CerSs), of which there are 6 different isoforms (CerS1 to -6)28 that generate ceramides with different fatty acid chain lengths.29 We found that treating PEL cell lines with the ceramide analogs upregulated the expression of CerS1, CerS3, and CerS4 (Figure 4G) and reduced the expression of the ceramide metabolizing enzymes, including SphK1, SphK2, and glucosylceramide synthase (GCS)30-32 (Figure 4H-I). Taken together, these results indicate that the ceramide analogs target both ceramide synthesizing and metabolizing enzymes leading to increased intracellular ceramide levels in PEL cells.
New ceramide analogs treatments suppress PEL progression in vivo
We first assessed the safe doses of analogs 315 and 403 in NOD/SCID mice. After drug treatment of 4 weeks, we found that 12.5 to 50 mg/kg were well tolerated by these mice, since no visible signs of cytotoxicity (eg, respiratory difficulties, feeding or drinking difficulties, spastic paralysis, convulsion or blindness, or weight lost) were seen during the treatment. At the end of treatment, the different organ tissues (liver, kidney, heart, lung, and spleen) were collected from the representative control, vehicle, or ceramide analog–treated mice. Histologic analysis (H&E staining) indicated no visible pathological changes in any of the organ tissues from ceramide analogs–treated mice when compared with the vehicle-treated (or control) mice (Figure 5A-B).
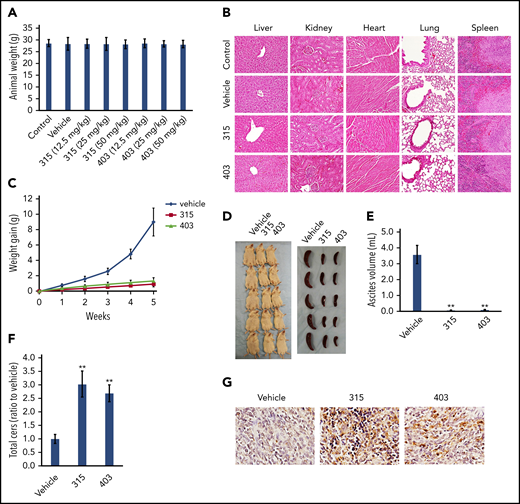
Treatment with the new ceramide analogs suppresses PEL progression with no visible cytotoxicity in vivo. (A) NOD/SCID mice (6- to 8 week old, male) were injected IP with 12.5, 25, 50 mg/kg of analog 315, 403, or vehicle (n = 5 per group), once daily, 2 days per week, and weights were recorded weekly. The average animal weights after the 4-week treatment period among different groups were compared. (B) At the end of the treatment period, the different organ tissues collected from the representative control, vehicle-, or ceramide analogs– (50 mg/kg) treated mice were observed and compared using the H&E staining (×60 magnification). (C-E) NOD/SCID mice were injected IP with 1 × 107 BCBL-1 cells. Twenty-four hours later, 25 mg/kg of analog 315, 403, or vehicle (n = 5 per group) was administered IP, once daily, 2 days per week, for each of the 2 independent experiments. Weights were recorded weekly. Images of the animals and their spleens as well as ascites fluid volumes were collected at the conclusion of the experiments after the 5-week treatment. Error bars represent SD for 1 of 2 independent experiments; **P < .01. (F) Total ceramide levels of ascites from each of 3 representative vehicle- or ceramide analog–treated mice were quantified using lipidomics analyses as described in the “Methods.” **P < .01. (G) Cell apoptosis in the ascites samples collected from the representative vehicle- or ceramide analog–treated mice was detected using the TUNEL assay (×60 magnification).
Treatment with the new ceramide analogs suppresses PEL progression with no visible cytotoxicity in vivo. (A) NOD/SCID mice (6- to 8 week old, male) were injected IP with 12.5, 25, 50 mg/kg of analog 315, 403, or vehicle (n = 5 per group), once daily, 2 days per week, and weights were recorded weekly. The average animal weights after the 4-week treatment period among different groups were compared. (B) At the end of the treatment period, the different organ tissues collected from the representative control, vehicle-, or ceramide analogs– (50 mg/kg) treated mice were observed and compared using the H&E staining (×60 magnification). (C-E) NOD/SCID mice were injected IP with 1 × 107 BCBL-1 cells. Twenty-four hours later, 25 mg/kg of analog 315, 403, or vehicle (n = 5 per group) was administered IP, once daily, 2 days per week, for each of the 2 independent experiments. Weights were recorded weekly. Images of the animals and their spleens as well as ascites fluid volumes were collected at the conclusion of the experiments after the 5-week treatment. Error bars represent SD for 1 of 2 independent experiments; **P < .01. (F) Total ceramide levels of ascites from each of 3 representative vehicle- or ceramide analog–treated mice were quantified using lipidomics analyses as described in the “Methods.” **P < .01. (G) Cell apoptosis in the ascites samples collected from the representative vehicle- or ceramide analog–treated mice was detected using the TUNEL assay (×60 magnification).
Next, we sought to determine whether these new ceramide analogs had the capacity to suppress PEL tumor growth in vivo using an established murine xenograft model wherein PEL cells were introduced into the peritoneal cavity of immunocompromised mice.33 We administered the ceramide analogs (or vehicle) IP within 24 hours of BCBL-1 injection and for 5 weeks of treatment. We found that treatment with either compounds dramatically suppressed PEL tumor progression, including reducing ascites formation and spleen enlargement over this timeframe (Figure 5C-E). The treatment of both compounds significantly increased total ceramides production from ascites of injected mice by using lipidomics analysis (Figure 5F). Furthermore, analysis of the TUNEL assays performed indicated significantly upregulated cell apoptosis in the ascites samples collected from the representative ceramide analog–treated mice when compared with the vehicle-treated mice (Figure 5G).
Transcriptomic analysis of gene profiles altered within new ceramide analog–treated PEL cells
To determine the overall metabolic changes induced by new ceramide analogs, we used the HumanHT-12 v4 Expression BeadChip (Illumina), which contains >47 000 probes derived from the NCBI RefSeq Release 38 and other sources to analyze the gene profile altered between vehicle- and 315- or 403-treated BCBL-1 and BCP-1 cells. Our analysis indicated that 59 and 52 genes were significantly changed (≥twofold, and P < .05) in 315-treated BCBL-1 and BCP-1 cells, respectively; 28 of those gene changes were changed similarly in these 2 cell lines (Figure 6A). There were 805 and 37 genes significantly changed in 403-treated BCBL-1 and BCP-1 cells (although it remains unknown why there is a big difference of gene profile changed between these 2 cell lines), respectively, and 34 of those gene changes were in common in the 2 cell lines. The top 8 commonly upregulated and downregulated candidate genes from 315- or 403-treated PEL cell lines are listed in supplemental Table 2 and supplemental Table 3, respectively. It is worth noting that only 2 “common” genes were downregulated in 403-treated BCBL-1 and BCP-1 cells. We next performed enrichment analysis of these significantly altered candidates by using the respective modules, including Pathway Maps, Gene Ontology Processes, and Process Networks obtained from Metacore Software. Not surprisingly, the top functional categories most candidates belong to include the regulation of lipid metabolism (eg, cholesterol biosynthesis), the regulation of different cell-cycle phases, and cytoskeleton/microtubules functions (Figure 6B-C; supplemental Figure 3 and -4). Notably, several cell proliferation-related signaling pathways, such as mTOR, MAPK, and NF-κB, were also affected by our ceramide analogs. We also performed a protein-protein interaction analysis for those commonly changed candidates in 315- or 403-treated PEL cell lines. Our results indicated 2 interaction clusters for the candidates from 315-treated PEL cells; in contrast, less protein interaction was observed in the candidates from 403-treated PEL cells (supplemental Figure 5).
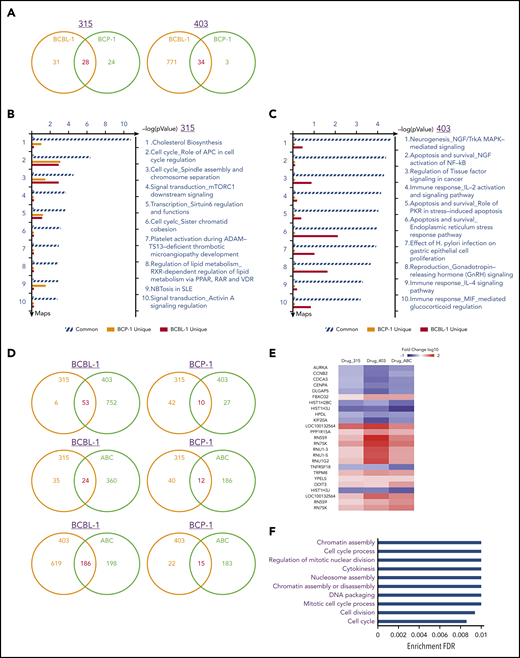
Comparative transcriptome analysis of gene profiles altered among new ceramide analogs and ABC294640 treated PEL cell lines. (A) The Human HT-12 v4 Expression BeadChip (Illumina) was used to investigate the transcriptome change between ceramide analogs (315 or 403) and vehicle-treated BCBL-1 and BCP-1 cells. The intersection analysis of significantly altered genes (expression change ≥ twofold; P < .05) was conducted using Illumina GenomeStudio Software. (B-C) The enrichment analysis of gene profile (common and unique) altered by ceramide analogs treatment was conducted using the Pathway Maps Module of MetaCore Software (Thompson Reuters). (D) The intersection analysis of significantly altered genes (expression change ≥ twofold; P < .05) was conducted as described above. (E-F) Heat map and enrichment analysis of genes commonly altered in new ceramide analogs and ABC294640 (ABC) treated PEL cell lines. IL-2, interleukin-2; MIF, macrophage migration inhibitory factor; NFG, nerve growth factor; PKR, protein kinase R; PPAR, peroxisome proliferator-activated receptors; RAR, retinoic acid receptor; SLE, systemic lupus erythematosus; VDR, vitamin D receptor.
Comparative transcriptome analysis of gene profiles altered among new ceramide analogs and ABC294640 treated PEL cell lines. (A) The Human HT-12 v4 Expression BeadChip (Illumina) was used to investigate the transcriptome change between ceramide analogs (315 or 403) and vehicle-treated BCBL-1 and BCP-1 cells. The intersection analysis of significantly altered genes (expression change ≥ twofold; P < .05) was conducted using Illumina GenomeStudio Software. (B-C) The enrichment analysis of gene profile (common and unique) altered by ceramide analogs treatment was conducted using the Pathway Maps Module of MetaCore Software (Thompson Reuters). (D) The intersection analysis of significantly altered genes (expression change ≥ twofold; P < .05) was conducted as described above. (E-F) Heat map and enrichment analysis of genes commonly altered in new ceramide analogs and ABC294640 (ABC) treated PEL cell lines. IL-2, interleukin-2; MIF, macrophage migration inhibitory factor; NFG, nerve growth factor; PKR, protein kinase R; PPAR, peroxisome proliferator-activated receptors; RAR, retinoic acid receptor; SLE, systemic lupus erythematosus; VDR, vitamin D receptor.
Comparative analysis of PEL gene profiles altered by new ceramide analogs and ABC294640
To further understand the influence of gene profiles in PEL cells treated with different sphingolipid metabolism-targeted molecules, we compared the transcriptomes among the cells treated with our 2 ceramide analogs and 1 specific sphingosine kinase inhibitor, ABC294640 treated. We found that only partial genes (53 in BCBL-1 and 10 in BCP-1) were commonly altered between 315- and 403-treated PEL cell lines (Figure 6D). The similar results were observed when comparing 315- or 403- to ABC294640-treated PEL cell lines (Figure 6D). These data confirm that these molecules differentially change the global gene expression in PEL cells, although they all induce intracellular ceramide production to kill tumor cells. For those commonly altered genes in PEL cells treated with any of these compounds (25 genes), we found that they displayed a consistent tendency of changes from the heat map (Figure 6E). Moreover, they belong to several functional categories affecting cell cycle, cell division, and chromatin assembly (Figure 6F), which are potentially controlled by sphingolipid metabolism and are responsible for PEL cell proliferation or survival. Our protein-protein interaction analysis indicated only 1 interaction cluster existed in these “common” candidates, which contains AURKA, CCNB2, CDCA3, CENPA, DLGAP5, HIST1H2BC, HIST1H3J, and KIF20A (supplemental Figure 6).
Identification of cellular genes required for PEL cell survival that are affected by the new ceramide analogs
Here, we selected 2 cellular candidates commonly changed in 315- and 403-treated PEL cells (actually changed in ABC294640-treated cells as well), AURKA (Aurora A kinase) and CDCA3 (cell division cycle-associated 3), to further validate their roles in PEL cell survival. Our qRT-PCR analysis confirmed both genes were significantly downregulated in 315- and 403-treated BCBL-1 cell lines (Figure 7A), similar to what we observed in microarray analysis. We next found that direct knockdown of either AURKA or CDCA3 by RNAi effectively repressed PEL cell growth through inducing lymphoma cell apoptosis (Figure 7B-E). Interestingly, we found that the expression of AURKA and CDCA3 were mainly controlled by SphK2 but not SphK1 in PEL cells (Figure 7F-G). Taken together, these data demonstrate that these cellular genes affected by our new ceramide analogs are indeed related to PEL cell survival and controlled by sphingolipid metabolism, which may serve as additional therapeutic targets.
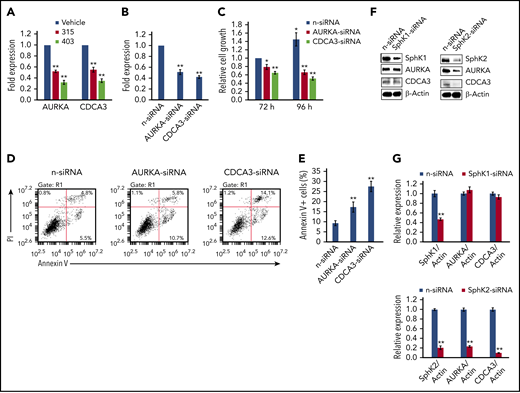
Cellular genes affected by new ceramide analogs are required for PEL cell survival. (A) The transcriptional levels of AURKA and CDCA3 were compared between ceramide analogs (315 or 403) and vehicle-treated BCBL-1 cells using qRT-PCR. (B-E) BCBL-1 was transfected with AURKA-siRNA, CDCA3-siRNA, or nontarget control siRNA (n-siRNA) for 72 to 96 hours; then the gene transcription, cell proliferation, and apoptosis were measured as described above. (F-G) BCBL-1 was transfected with SphK1-siRNA, SphK2-siRNA, or n-siRNA for 72 hours; then the protein expression was measured as described above. Error bars represent SD for 3 independent experiments. *P < .05; **P < .01.
Cellular genes affected by new ceramide analogs are required for PEL cell survival. (A) The transcriptional levels of AURKA and CDCA3 were compared between ceramide analogs (315 or 403) and vehicle-treated BCBL-1 cells using qRT-PCR. (B-E) BCBL-1 was transfected with AURKA-siRNA, CDCA3-siRNA, or nontarget control siRNA (n-siRNA) for 72 to 96 hours; then the gene transcription, cell proliferation, and apoptosis were measured as described above. (F-G) BCBL-1 was transfected with SphK1-siRNA, SphK2-siRNA, or n-siRNA for 72 hours; then the protein expression was measured as described above. Error bars represent SD for 3 independent experiments. *P < .05; **P < .01.
Discussion
After screening 19 newly synthesized ceramide analogs (Figure 1A), we have identified 6 compounds that display the most effective anti-PEL activities. Based on the structure, these 6 compounds can be divided into 3 subgroups: (1) analogs 301, 309, 313, and 315 all have a serine-derived backbone with an amide functionality, and an imine functional group with a substituted phenyl ring in the side chain. These compounds differ in the type and location of the phenyl ring substituents: 301 has a nitro group at the para position; 309 has a chlorine at the ortho position; 313 has a hydroxyl group at the para position; and 315 has a hydroxyl group at the ortho position. (2) Analog 403 has a 3-hydroxy-4-ene backbone structure similar to naturally occurring ceramide, with a modified side chain containing a phenylacetyl amide. (3) Analog 953 has an amide functionality in the long carbon chain backbone, and a sulfonamide functionality in the side chain containing a 4-methyl–substituted phenyl group. We then selected analogs 315 and 403, both of which displayed effective anti-PEL activity in a dose-dependent manner for the subsequent experiments. Notably, analog 315 contains an imine functional group and a phenolic hydroxyl group, which increase permeability through the lipid bilayer as well as water solubility. More importantly, both compounds showed IC50 ranges in PEL cell lines that are much lower than those of other sphingolipid metabolism-targeted molecules, especially those of natural short-/long-chain ceramides (Figure 2G). These comparative data strongly demonstrate the advantage of our new ceramide analogs in therapeutic efficacy for PEL tumors. Interestingly, both compounds also have been found with anticancer activities on some chemoresistant breast cancer cells.23,34,35 In further studying drug selectivity, we found that analog 403 showed inhibitory effects on the growth of diffuse large B-cell lymphoma cell lines, such as Su-DHL-6 and HBL-1 (supplemental Figure 7A-B), while analog 315 did not (data not shown). PEL cells are characterized by hallmarks of plasma cell differentiation, while multiple myeloma represents another tumor entity from plasma cells. In these studies, we found that both analogs 315 and 403 showed much less inhibitory effects on the growth of multiple myeloma cell lines, RPMI-8266, and U266 (only partially effective at 40 μM concentration; supplemental Figure 7C-D).
In addition to the regulation of intracellular ceramide concentrations, sphingolipid metabolism also regulates many other cellular factors, which determine cell survival/death. Our recent transcriptomic analyses revealed that a subset of tumor suppressor genes (∼25 genes) was significantly upregulated in KSHV+ PEL cell lines by exogenous dhC16-Cer.19 Among these tumor suppressor genes, S100A2 and THBS1 have been reported as direct cellular targets by multiple KSHV-microRNAs.36,37 Repression of THBS1 expression also reduces downstream transforming growth factor-β signaling activities,36 which are involved in enhancing cell survival and angiogenesis in KSHV-infected cells.38,39 Silencing THBS1 by RNAi partially rescued PEL cells from dhC16-Cer-induced cell-cycle arrest.19 In the current study, we have identified a subset of cellular genes affected by our new ceramide analogs using microarray analysis, and some of them (eg, AURKA and CDCA3) are indeed functionally related to PEL cell survival that have never been reported. Interestingly, 1 recent study has found that inhibition of AURKA in combination with chemotherapy induces synthetic lethality and overcomes chemoresistance in MYC-overexpressing lymphoma, including non-Hodgkin lymphoma.40 Dauch et al recently demonstrated a direct binding of AURKA to MYC, which results in the stabilization of MYC protein.41 Interestingly, our recent data showed that treatment with either analog 315 or 403 significantly reduced MYC expression from PEL cells (supplemental Figure 8). Moreover, AURKA-targeted therapies have been recently developed for adult T-cell leukemia/lymphoma (especially MLN8237, also known as alisertib, 1 selective AURKA inhibitor).42-45 Although unknown for its functional role in lymphoma, CDCA3 has been identified as a prognostic biomarker and potential therapeutic target in multiple cancers, such as prostate, lung, bladder, colorectal, and gastric cancer.46-50 We also found that the expression of AURKA and CDCA3 were controlled by SphK2 but not SphK1, although the underlying mechanisms still need further investigation. Our overall findings indicate that the sphingolipid metabolism may have a broader role in the regulation of cellular factors in cancer cells, such as PEL.
We also investigated whether treatment with our new ceramide analogs affected KSHV viral gene profile in PEL, BCBL-1, and BCP-1 cell lines (supplemental Figure 9). By using qRT-PCR, we found that both 315 and 403 significantly increased lytic gene expression, such as RTA (Immediate early gene), ORF59 (Early gene), and ORF17 (Late gene), but did not change latent gene expression, such as LANA. Notably, analog 403 induced an intensive increase in lytic gene expression in BCBL-1 cells, which may explain why treatment with this compound caused a larger number of cellular gene alterations in BCBL-1 cells in comparison with other cell lines (Figure 6A). Our data demonstrate the tight connection between sphingolipid metabolism and KSHV gene expression in PEL cells. In many cases, PEL cells are coinfected by EBV, another oncogenic human γ-herpesvirus.51,52 Therefore, we detected whether treatment with our new ceramide analogs may affect EBV gene profile in KSHV+/EBV+ PEL cell lines. Interestingly, our results indicated that both analogs 315 and 403 significantly reduced EBV lytic gene expression (eg, BHRF1 and BZLF1), while having little effect on latent gene expression (eg, EBNA1) from JSC-1 and BC-1 cell lines (supplemental Figure 10). Our data imply that these new compounds (or sphingolipid metabolism) may differentially regulate KSHV and EBV gene profiles within PEL cells.
In the current study, we have found that treatment with analog 315 or 403 effectively suppresses PEL progression in immunocompromised mice, while showing no obvious toxicity at the doses we used. The study reported here only focuses on single-agent treatment of PEL cells. In fact, recent studies have reported synergistic effects by using the combinations of chemotherapeutic drugs and ceramide-generating agents (eg, CerSs or SMase inducers) or modulators of ceramide metabolism (eg, GCS or SphKs inhibitors).53 Interestingly, evidence also shows a close relationship between GCS and P-glycoprotein (an important protein involved in multidrug-resistance). Overexpression of P-glycoprotein in resistant cancer cells is concomitant with high GCS expression.54 Some ceramide inducers have been shown to sensitize cancer cells to anticancer agents, resulting in enhanced cell apoptosis and death53 ; however, such effects need to be further explored in our PEL xenograft models. Published data have shown that PEL cells are differentially resistant to doxorubicin, and both p53 mutagenesis and KSHV-encoded LANA-2 protein induced chemoresistance for KSHV+ lymphoma.55,56 Our recent study has demonstrated that the multifunctional glycoprotein, Emmprin, interacts with the lymphatic vessel endothelial hyaluronan receptor-1 and a drug transporter known as the breast cancer resistance protein to facilitate PEL chemoresistance to doxorubicin and paclitaxel.25 Therefore, it will be interesting to determine whether treatment with our ceramide analogs would interfere with the expression or function of these chemoresistance-related factors to reduce chemoresistance in PEL cells.
Although our new ceramide analogs show promising therapeutic applications, there are limitations extrapolating from the current study. For example, it is highly desirable to use orally effective drugs for treating PEL; however, the efficacy of the novel analogs in the PEL xenograft model following oral administration has not been explored and thus remains unclear. Another limitation relates to the possibility of tumor resistance and recurrence during or after treating PEL cells with these new ceramide analogs, and if resistance or recurrence develops, how will they be overcome? Finally, although the ceramide analogs show good efficacy with micromolar potencies, increasing their potency by additional chemical modification may further increase their specificity and ability to treat PEL.
The microarray original data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE137123).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by National Institutes of Health (NIH), National Cancer Institute grant R01CA228166 (Z.Q.). Support has also been provided in part by the Arkansas Bioscience Institute, the major research component of the Arkansas Tobacco Settlement Proceeds Act of 2000. This work was also supported by an NIH COBRE grant P20 GM121288, a Tulane school of medicine faculty research pilot grant, and a Carol Lavin Bernick faculty grant (Z.L.). The Foroozesh group acknowledges the funding by the DoD Breast Cancer grant W81XWH-11-1-0105, the NIH award R15CA159059, National Institute on Minority Health and Health Disparities award G12MD007595, and the Louisiana Cancer Research Center.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Authorship
Contribution: L.D., N.G., S.R.P., M.F., and Z.Q. designed and performed experiments, analyzed results, and wrote the manuscript; M.F. and Z.Q. are the corresponding authors; J.C., L.D.V., and J.Z. performed the experiments; and J.C., Z.L., and J.L. performed statistical and microarray analysis or provided critical input.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Zhiqiang Qin, University of Arkansas for Medical Sciences, 4301 W Markham St, Little Rock, AR 72205; e-mail: zqin@uams.edu; and Maryam Foroozesh, Xavier University of Louisiana, 1 Drexel Dr, New Orleans, LA 70125; e-mail: mforooze@xula.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal