

Key Points
This largest study to date of CANOMAD patients revealed that one third harbored an overt hematologic malignancy, consisting mainly in WM.
IVIg and rituximab-based regimens were the most effective therapies with a 50% response rate.

Abstract
CANOMAD (chronic ataxic neuropathy, ophthalmoplegia, immunoglobulin M [IgM] paraprotein, cold agglutinins, and disialosyl antibodies) is a rare syndrome characterized by chronic neuropathy with sensory ataxia, ocular, and/or bulbar motor weakness in the presence of a monoclonal IgM reacting against gangliosides containing disialosyl epitopes. Data regarding associated hematologic malignancies and effective therapies in CANOMAD are scarce. We conducted a French multicenter retrospective study that included 45 patients with serum IgM antibodies reacting against disialosyl epitopes in the context of evocating neurologic symptoms. The main clinical features were sensitive symptoms (ataxia, paresthesia, hypoesthesia; n = 45, 100%), motor weakness (n = 18, 40%), ophthalmoplegia (n = 20, 45%), and bulbar symptoms (n = 6, 13%). Forty-five percent of the cohort had moderate to severe disability (modified Rankin score, 3-5). Cold agglutinins were identified in 15 (34%) patients. Electrophysiologic studies showed a demyelinating or axonal pattern in, respectively, 60% and 27% of cases. All patients had serum monoclonal IgM gammopathy (median, 2.6 g/L; range, 0.1-40 g/L). Overt hematologic malignancies were diagnosed in 16 patients (36%), with the most frequent being Waldenström macroglobulinemia (n = 9, 20%). Forty-one patients (91%) required treatment of CANOMAD. Intravenous immunoglobulins (IVIg) and rituximab-based regimens were the most effective therapies with, respectively, 53% and 52% of partial or better clinical responses. Corticosteroids and immunosuppressive drugs were largely ineffective. Although more studies are warranted to better define the optimal therapeutic sequence, IVIg should be proposed as the standard of care for first-line treatment and rituximab-based regimens for second-line treatment. These compiled data argue for CANOMAD to be included in neurologic monoclonal gammopathy of clinical significance.
Medscape Continuing Medical Education online
In support of improving patient care, this activity has been planned and implemented by Medscape, LLC and the American Society of Hematology. Medscape, LLC is jointly accredited by the Accreditation Council for Continuing Medical Education (ACCME), the Accreditation Council for Pharmacy Education (ACPE), and the American Nurses Credentialing Center (ANCC), to provide continuing education for the healthcare team.
Medscape, LLC designates this Journal-based CME activity for a maximum of 1.00 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Successful completion of this CME activity, which includes participation in the evaluation component, enables the participant to earn up to 1.0 MOC points in the American Board of Internal Medicine's (ABIM) Maintenance of Certification (MOC) program. Participants will earn MOC points equivalent to the amount of CME credits claimed for the activity. It is the CME activity provider's responsibility to submit participant completion information to ACCME for the purpose of granting ABIM MOC credit.
All other clinicians completing this activity will be issued a certificate of participation. To participate in this journal CME activity: (1) review the learning objectives and author disclosures; (2) study the education content; (3) take the post-test with a 75% minimum passing score and complete the evaluation at http://www.medscape.org/journal/blood; and (4) view/print certificate. For CME questions, see page 2481.
Disclosures
Associate Editor Laurie H. Sehn served as advisor or consultant for AbbVie Inc., Acerta Pharma, Amgen Inc., Apobiologix, AstraZeneca Pharmaceuticals LP, Celgene Corporation, Gilead Sciences, Inc., H. Lundbeck A/S, Janssen Pharmaceuticals, Inc., Karyopharm Therapeutics, Kite Pharma Inc., Merck & Co., Inc., MorphoSys, Roche/Genentech, Inc., SeaGen Inc., Takeda, Teva, TG Therapeutics, Inc., and Verastem Oncology and received grants for clinical research from H. Lundbeck A/S. Author Véronique Leblond received honoraria and advisory board fees from Roche. CME questions author Laurie Barclay, freelance writer and reviewer, Medscape, LLC and the remaining authors declare no competing financial interests.
Learning objectives
Upon completion of this activity, participants will be able to:
Describe clinical features of chronic ataxic neuropathy, ophthalmoplegia, immunoglobulin M paraprotein, cold agglutinins and disialosyl antibodies (CANOMAD)/chronic ataxic neuropathy with disialosyl antibodies (CANDA) syndrome, according to a French multicenter retrospective study
Determine hematologic malignancies associated with CANOMAD/CANDA syndrome, according to a French multicenter retrospective study
Identify optimal treatment of CANOMAD/CANDA syndrome, according to a French multicenter retrospective study
Release date: November 19, 2020; Expiration date: November 19, 2021
Introduction
Monoclonal gammopathy of unknown significance is a common age-related condition, defined by the presence in serum and/or urine of a monoclonal immunoglobulin produced by an abnormal B-cell clone, in the absence of overt plasma cell or B-cell lymphoproliferative diseases. Although mainly asymptomatic, these quiescent clones can be responsible for potentially severe organ damage because of the toxicity of the monoclonal immunoglobulin or other mechanisms, defining monoclonal gammopathy of clinical significance (MGCS).1 This situation often requires therapeutic intervention, directed against the B-cell clone.2 Along with the kidney and skin, the peripheral nerve is one of the most frequently involved organs.
Monoclonal gammopathies associated with peripheral neuropathy are more commonly immunoglobulin M (IgM) than IgG or IgA. The prevalence of neuropathy in patients with monoclonal IgM ranges from 5% to 31%.3,4 Pathophysiologic mechanisms that link gammopathy and neuropathy include (1) specific autoantibody activity of the IgM against different components of the nerve, (2) specific properties of circulating IgM, leading to cryoglobulinemic neuropathy, amyloid, or endoneurial IgM deposits, and (3) nerve infiltration or nerve damage mediated by cytokine secretion of tumoral cells.1,5 Autoreactive IgM most frequently recognize myelin-associated glycoprotein (MAG), but gangliosides can also be targeted. This latter activity can be responsible for a neurologic syndrome termed CANOMAD (chronic ataxic neuropathy, ophthalmoplegia, IgM paraprotein, cold agglutinins and disialosyl antibodies), which is characterized by chronic neuropathy with sensory ataxia and motor weakness involving oculomotor and bulbar muscles, occurring in the presence of monoclonal IgM directed against disialosyl ganglioside epitopes. As some of the symptoms defining CANOMAD are not observed in all patients,6,7 this acronym may be restrictive, and the term CANDA (chronic ataxic neuropathy with disialosyl antibodies) has also been proposed.8 The first case of neuropathy associated with monoclonal IgM with anti-ganglioside auto-activity was described in 1985,9 and the largest series to date, published in 2001, comprised 18 patients.6 Since then, several case reports and small series have been reported in the literature, amounting to less than 50 cases.6,9-18 Symptoms are dominated by progressive chronic neuropathy with marked sensory ataxia that can lead to severe disability. In some cases, fluctuating or fixed symptoms such as ocular, sensory, or bulbar manifestations are reported together with ataxia. The presence of antibodies directed against gangliosides is constant. The most frequently targeted gangliosides are GD1b, GD3, GT1b, and GQ1b, which all harbor disialosyl groups containing the sequence NeuNAc (α2-8) NeuNAc (α2-3) Gal. Such gangliosides are notably localized in the neurons of dorsal root ganglia and within the oculomotor nerves.19 In contrast to anti-MAG neuropathy, which is a predominantly distal demyelinating neuropathy, both axonal and demyelinating patterns have been reported using electrophysiologic studies and nerve biopsies.
Investigation of IgM-associated peripheral neuropathy requires a well-defined strategy that combine (1) nerve conduction studies (NCSs) to distinguish between demyelinating and axonal patterns; (2) detection of anti-MAG and antiganglioside antibodies; and (3) screening for “red flag” features (that include pain, multifocal topography, rapidly evolving course, cranial nerve involvement, dysautonomia, weight loss, cutaneous signs, heart/kidney/lung involvement, and abnormal serum-free light-chain concentration and ratio) to orientate further investigations and rule out in particular cryoglobulinemic/amyloid neuropathy and tumor nerve infiltration.5,20
There is actually no consensus for CANOMAD/CANDA treatment. Different therapeutic options have included intravenous immunoglobulins (IVIg), plasma exchange, corticosteroids, and immunosuppressive drugs.7 More recently, small case series have reported encouraging results with rituximab.21-24 Finally, although virtually all cases of CANOMAD/CANDA syndrome are associated with the presence of a monoclonal IgM, the spectrum of underlying hematologic malignancies and the efficacy of B-cell–targeting therapies have not been thoroughly investigated.
The main objectives of this retrospective study were to describe the clinical features of CANOMAD/CANDA syndrome, to identify associated hematologic malignancies, and to gain insights into its optimal therapeutic management while studying the largest patient cohort to date.
Methods
Patients
This study is a national retrospective analysis of 45 patients, treated in 17 centers, which were included in a CANOMAD multicenter French registry between 2002 and 2018 on the basis of the positivity of at least 1 serum IgM antibody reacting against disialosyl epitopes (among GD1b, GD3, GT1b, and GQ1b) and evocating neurologic signs. A standardized questionnaire on demographics, clinical, biological, electrophysiologic, and therapeutic data were sent to all referring clinicians.
IVIg treatment consisted of a dose of 2 g/kg every 4 weeks. This interval between courses could be gradually increased by 1 week in case of clinical improvement. IVIg treatment could be stopped in case of clinical complete response (CR). Administration schedules of single-agent rituximab were weekly infusions of 375 mg/m2 for 4 weeks (n = 13) or 2 infusions of 1 g/m2 on days 1 and 15 (n = 6).
Clinical data were obtained in accordance with the Declaration of Helsinki and its later amendments. All patients were informed, and they consented to participate in this study before inclusion in the registry.
Three cases have been published previously as case reports.21,25,26
Investigations
Detection of monoclonal gammopathies, cold agglutinins, and antiganglioside antibodies were performed locally and were part of variables collected in the standardized questionnaire. Methods testing for the presence of antidisialosyl antibodies comprised enzyme-linked immunosorbent assay,6,27 “home-made” immunodots,28 or commercial tests (Generic Assays, Les Ulis, Courtaboeuf, France). The Generic Assays commercial test (AntiGangliosidDot) is a line immunoassay used for the qualitative detection of IgM antibodies directed against 11 gangliosides (GM1, GM2, GM3, GM4, GD1a, GD1b, GD2, GD3, GT1a, GT1b, and GQ1b), whereas the home-made assay detect antibodies directed against 8 gangliosides (GM1, GM2, GM3, GD1a, GD1b, GD2, GT1b, GQ1b). Routine screening investigations to exclude other causes of neuropathy followed recommendations previously published5 and were negative. Electrophysiologic studies were carried out locally in all cases and centrally reviewed. Axonal and/or demyelinating damages were defined according to the Ad Hoc Subcommittee of the American Academy of Neurology AIDS Task Force’s demyelination criteria.29 Bone marrow examination was performed in 31 of 45 (69%) patients.
Definitions
CANOMAD and CANDA were defined as described in the Introduction section and in previous publications.6,8 Diagnoses of associated hematologic malignancies followed the World Health Organization classification criteria.30
To assess treatment response, we used the modified Rankin score (RS) (a 7-point disability rating scale with scores from 0 [no deficit] to 6 [death]) evaluating the degree of disability or dependence for daily activities.31 Clinical response was defined as CR (complete clinical improvement from baseline, RS = 0), partial response (PR; a clinical improvement defined as ≥1-point RS decrease), stable disease (SD; unchanged RS), and progression (≥1-point RS increase). The different lines of therapy were separately analyzed for each patient. Biological response was defined as CR (disappearance of IgM and negative immunofixation), PR (decrease in serum IgM levels >50%), SD (increase <25% or decrease <25%), and progression (increase >25%).
Statistics
Overall survival was calculated from date of diagnosis until last follow-up/death and time to next treatment from date of first to second line of treatment. Kaplan-Meier analysis was performed to construct survival curves, and the log-rank test was used to determine differences between groups. The χ2 test or Fisher’s exact test was used to compare data distribution in different clinical or treatment subgroups. The significance level of P < .05 was applied, and statistical analyses were performed using SPSS software.
Results
Clinical features
The main clinical characteristics are provided in Table 1. Of the 45 patients included in our study, 35 (78%) were male. Median age at onset of symptoms was 58 years (range, 37-81 years), and the median delay between onset of symptoms and diagnosis of CANOMAD/CANDA was 4.0 years (range, 0.1-27 years). The most frequent initial symptoms were sensory (paresthesia and ataxia in respectively 26 [58%] and 21 [47%] cases), ocular (n = 6, 13%), and bulbar (n = 3, 7%). Of note, the disease was revealed by isolated ophthalmoplegia in 1 patient. At CANOMAD/CANDA diagnosis, nearly half (n = 19, 44%) of the cohort harbored significant disability (modified RS of 3-5). Clinical outcome was mainly chronic progressive (n = 30, 67%) or relapsing-remitting (n = 14, 31%). One patient experienced 1 symptomatic flare-up that completely resolved after first-line therapy. The set of neurologic symptoms observed during the course of the disease for the whole cohort is detailed in Table 1.
Main clinical and electrophysiologic characteristics of CANOMAD/CANDA patients
| Characteristics | No. (%) or median (range) |
|---|---|
| Total | 45 (100) |
| Male | 35 (78) |
| Age at CANOMAD/CANDA diagnosis, y | |
| Median (range) | 62 (38-81) |
| Time from onset of symptoms to CANOMAD/CANDA diagnosis, y | |
| Median (range) | 4 (0.1-27) |
| Type of onset | |
| Acute | 8 (18) |
| Subacute (rapidly progressive) | 7 (15) |
| Chronic (slowly progressive) | 30 (67) |
| Neurologic symptoms at diagnosis | |
| Sensory symptoms | 35 (78) |
| Paresthesia | 26 (58) |
| Ataxia | 21 (47) |
| Ophthalmoplegia | 6 (13) |
| Bulbar symptoms | 3 (7) |
| Others | 14 (30) |
| Painful limb | 6 (13) |
| Motor weakness/myoclonus | 3 (7) |
| Acrocyanosis/livedo | 2 (4) |
| Dyspnea | 1 (2) |
| Facial nerve paralysis | 2 (4) |
| Neurologic symptoms during the course of the disease | |
| Sensory ataxia | 34 (76) |
| Paresthesia | 35 (78) |
| Hypoesthesia | 42 (93) |
| Upper and lower limbs | 24 (53) |
| Lower limbs only | 18 (40) |
| Ophthalmoplegia | 20 (44) |
| Bulbar symptoms | 6 (13) |
| Motor weakness | 18 (40) |
| Upper and lower limbs | 13 (29) |
| Lower limbs only | 5 (11) |
| Areflexia | 42 (93) |
| Facial nerve paralysis | 3 (7) |
| Dysautonomic signs | 2 (4) |
| Acute respiratory distress | 3 (7) |
| Modified Rankin score at diagnosis | /42 |
| 0 (asymptomatic) | 0 (0) |
| 1 (symptomatic but no significant disability) | 15 (36) |
| 2 (slight disability) | 8 (19) |
| 3 (moderate disability) | 7 (17) |
| 4 (moderately severe disability) | 10 (24) |
| 5 (severe disability) | 2 (5) |
| Electrophysiologic findings, pattern | |
| Demyelinating | 27 (60) |
| Axonal | 12 (27) |
| Normal | 6 (3) |
| Type of evolution | |
| Relapsing-remitting | 14 (31) |
| Chronic progressive | 30 (67) |
| Isolated symptomatic flare-up | 1 (2) |
| Characteristics | No. (%) or median (range) |
|---|---|
| Total | 45 (100) |
| Male | 35 (78) |
| Age at CANOMAD/CANDA diagnosis, y | |
| Median (range) | 62 (38-81) |
| Time from onset of symptoms to CANOMAD/CANDA diagnosis, y | |
| Median (range) | 4 (0.1-27) |
| Type of onset | |
| Acute | 8 (18) |
| Subacute (rapidly progressive) | 7 (15) |
| Chronic (slowly progressive) | 30 (67) |
| Neurologic symptoms at diagnosis | |
| Sensory symptoms | 35 (78) |
| Paresthesia | 26 (58) |
| Ataxia | 21 (47) |
| Ophthalmoplegia | 6 (13) |
| Bulbar symptoms | 3 (7) |
| Others | 14 (30) |
| Painful limb | 6 (13) |
| Motor weakness/myoclonus | 3 (7) |
| Acrocyanosis/livedo | 2 (4) |
| Dyspnea | 1 (2) |
| Facial nerve paralysis | 2 (4) |
| Neurologic symptoms during the course of the disease | |
| Sensory ataxia | 34 (76) |
| Paresthesia | 35 (78) |
| Hypoesthesia | 42 (93) |
| Upper and lower limbs | 24 (53) |
| Lower limbs only | 18 (40) |
| Ophthalmoplegia | 20 (44) |
| Bulbar symptoms | 6 (13) |
| Motor weakness | 18 (40) |
| Upper and lower limbs | 13 (29) |
| Lower limbs only | 5 (11) |
| Areflexia | 42 (93) |
| Facial nerve paralysis | 3 (7) |
| Dysautonomic signs | 2 (4) |
| Acute respiratory distress | 3 (7) |
| Modified Rankin score at diagnosis | /42 |
| 0 (asymptomatic) | 0 (0) |
| 1 (symptomatic but no significant disability) | 15 (36) |
| 2 (slight disability) | 8 (19) |
| 3 (moderate disability) | 7 (17) |
| 4 (moderately severe disability) | 10 (24) |
| 5 (severe disability) | 2 (5) |
| Electrophysiologic findings, pattern | |
| Demyelinating | 27 (60) |
| Axonal | 12 (27) |
| Normal | 6 (3) |
| Type of evolution | |
| Relapsing-remitting | 14 (31) |
| Chronic progressive | 30 (67) |
| Isolated symptomatic flare-up | 1 (2) |
Serologic analyses and other investigations
All patients had antiganglioside antibodies with IgM specificity, reacting against at least 1 of 4 of the following disialosyl epitopes: GD1b, GD3, GT1b, and GQ1b. Thirty (67%) and 21 (47%) patients had antibodies reacting against at least 3 or 4 of 4 of the mentioned epitopes. Forty (89%) had also antibodies reacting against other gangliosides such as GM1, GM2, GM3, GD1a, GD2, or GT1a.
Regarding cerebrospinal fluid analysis, the mean protein level was 0.9 g/L (range, 0-2 g/L), and cellularity was negative in 80% of cases (median, 0/mm3; range, 0-4/mm3). NCSs showed a demyelinating or axonal pattern in, respectively, 27 (60%) and 12 patients (27%). Among the 6 patients for whom electrophysiologic studies were normal, 3 had abnormal somatosensory evoked potential, corresponding to a particular form of peripheral neuropathy, characterized by demyelination involving the proximal part of the sensory nerves32 ; it could be undetectable by classical NCSs. One patient had a form of small fiber neuropathy, which is also usually not detected by classical NCSs. Two other patients had only very mild sensitive symptoms when NCSs were performed. Fourteen nerve biopsies were performed revealing demyelinating, axonal, and mixed features in 4, 5, and 4 cases, respectively. One patient had a normal nerve biopsy. Three cases presented additional abnormalities: endoneural fibrosis (n = 1), IgM deposits identified by immunofluorescence (n = 1), and epineural lymphocyte infiltrate without precision (n = 1).
Associated hematologic and nonhematologic malignancies
All patients (n = 45) harbored a monoclonal IgM gammopathy detected by immunofixation. The monoclonal light chain was κ in 19 of 45 (42%) and λ in 21 of 45 (49%) patients. Five patients had both monotypic κ and λ IgM. The median peak value was 2.6 g/L (range, 0.1-40 g/L). Cold agglutinins were identified in 15 of the 44 screened cases (34%), but their specificity was not available in most cases. None had active hemolysis.
Overt associated hematologic malignancies were identified in 16 patients (36%). These diagnoses were made before, after, or concomitant to those of CANOMAD/CANDA in 4 (25%), 4 (25%), and 8 (50%) patients, respectively. One patient required treatment of B-cell malignancy before CANOMAD/CANDA diagnosis. The most frequent associated hematologic malignancy was Waldenström macroglobulinemia (WM; n = 9, 20%). Other documented hematologic neoplasms included diffuse large B-cell lymphoma (n = 2, 4%, including 1 transformation of WM), monoclonal B lymphocytosis (n = 2, 4%), chronic lymphocytic leukemia (n = 1, 2%), marginal zone lymphoma (n = 1, 2%), unclassifiable small B-cell lymphoma (n = 1, 2%), and mantle cell lymphoma (n = 1, 2%; Table 2). Clinical characteristics of CANOMAD/CANDA were largely similar between patients with or without an overt associated hematologic malignancy, notably in terms of ocular or bulbar involvement and degree of disability, and were no significantly associated with the level of IgM peak value (supplemental Tables 1, 1bis, and 1ter, available on the Blood Web site).
Hematologic neoplasms associated with CANOMAD/CANDA syndrome
| No. (%) | |
|---|---|
| Monoclonal gammopathy | 45 (100) |
| IgM | 45 (100) |
| κ | 19 (42) |
| λ | 21 (49) |
| κ and λ | 5 (11) |
| Associated hematologic malignancies | 17 (38) |
| Waldenström macroglobulinemia | 9 (20) |
| Diffuse large B-cell lymphoma | 2 (4) |
| Monoclonal B lymphocytosis | 2 (4) |
| Marginal zone lymphoma | 1 (2) |
| Chronic lymphocytic leukemia | 1 (2) |
| Unclassifiable small B-cell lymphoma | 1 (2) |
| Mantle cell lymphoma | 1 (2) |
| No. (%) | |
|---|---|
| Monoclonal gammopathy | 45 (100) |
| IgM | 45 (100) |
| κ | 19 (42) |
| λ | 21 (49) |
| κ and λ | 5 (11) |
| Associated hematologic malignancies | 17 (38) |
| Waldenström macroglobulinemia | 9 (20) |
| Diffuse large B-cell lymphoma | 2 (4) |
| Monoclonal B lymphocytosis | 2 (4) |
| Marginal zone lymphoma | 1 (2) |
| Chronic lymphocytic leukemia | 1 (2) |
| Unclassifiable small B-cell lymphoma | 1 (2) |
| Mantle cell lymphoma | 1 (2) |
Three patients developed solid tumors during their follow-up: bronchopulmonary cancer (n = 2) and glioblastoma (n = 1). These diagnoses were concomitant in 1 case and largely posterior to those of CANOMAD/CANDA in 2 cases (6 and 18 years).
Treatment and outcomes
Forty-one (91%) patients received treatment of CANOMAD/CANDA, including 2 patients treated by CHOP (cyclophosphamide, hydroxydaunorubicin, vincristine [Oncovin], prednisone) regimen for concomitant symptomatic hematologic malignancy. Among the 4 patients that did not receive treatment, 3 had minor symptoms (modified RS of 1) and a very indolent course with a follow-up of more than 5 years each. One patient had a very aggressive and disabling CANOMAD syndrome revealed by acute respiratory distress secondary to phrenic paralysis and died within a few weeks because of pulmonary complications.
The median number of therapeutic lines was 2 (mean, 3.0; range, 1-7), corresponding to a cumulative number of 135 lines of treatment of the whole cohort. The median delay between CANOMAD/CANDA diagnosis and initiation of treatment was 4 months (range, 0-168). First-line treatment consisted in IVIg (n = 20/41, 49%), corticosteroids (n = 11/41, 27%), plasma exchange (n = 3/41, 7%), chlorambucil (n = 3/41, 7%), CHOP regimen (n = 2/41, 5%), azathioprine (n = 1/41, 2%), or rituximab (n = 1/41, 2%; Table 3). Of note, clinical characteristics, especially regarding the degree of disability before treatment, were similar between the 2 largest groups of first-line treatment (IVIg and corticosteroids; data not shown). Overall clinical response was obtained in 18 (44%) patients, including 4 (10%) CR and 14 (34%) PR, whereas SD and progression were, respectively, observed in 7 (17%) and 16 (39%) cases (Table 3; supplemental Figure 1). Type of first-line treatment impacted quality of response because overall clinical responses were obtained in 60% of patients treated by IVIg and in only 1 patient (10%) with corticosteroids, correlating with a respective median decrease of 1 point (range, +1 to −4) and increase of 1 point (range, 0 to +2) in modified RS.
First-line therapies and clinical responses
| First-line | n | Clinical responses, n (%) | |||
|---|---|---|---|---|---|
| CR | PR | SD | Progression | ||
| IVIg | 20 | 4 (20) | 8 (40) | 6 (30) | 2 (10) |
| Corticosteroids | 11 | 0 (0) | 1 (10) | 0 (0) | 10 (90) |
| Chlorambucil | 3 | 0 (0) | 1 (33) | 0 (0) | 2 (67) |
| Plasma exchange | 3 | 0 (0) | 2 (67) | 0 (0) | 1 (33) |
| CHOP regimen | 2 | 0 (0) | 1 (50) | 0 (0) | 1 (50) |
| Azathioprine | 1 | 0 (0) | 0 (0) | 1 (100) | 0 (0) |
| Rituximab | 1 | 0 (0) | 1 (100) | 0 (0) | 0 (0) |
| Total | 41 | 4 (10) | 14 (34) | 7 (17) | 16 (39) |
| First-line | n | Clinical responses, n (%) | |||
|---|---|---|---|---|---|
| CR | PR | SD | Progression | ||
| IVIg | 20 | 4 (20) | 8 (40) | 6 (30) | 2 (10) |
| Corticosteroids | 11 | 0 (0) | 1 (10) | 0 (0) | 10 (90) |
| Chlorambucil | 3 | 0 (0) | 1 (33) | 0 (0) | 2 (67) |
| Plasma exchange | 3 | 0 (0) | 2 (67) | 0 (0) | 1 (33) |
| CHOP regimen | 2 | 0 (0) | 1 (50) | 0 (0) | 1 (50) |
| Azathioprine | 1 | 0 (0) | 0 (0) | 1 (100) | 0 (0) |
| Rituximab | 1 | 0 (0) | 1 (100) | 0 (0) | 0 (0) |
| Total | 41 | 4 (10) | 14 (34) | 7 (17) | 16 (39) |
Thirteen patients (32%) received only 1 line of treatment, which consisted of IVIg (n = 10), single-agent rituximab (n = 1), plasma exchange (n = 1), and CHOP regimen (n = 1). Median time to next treatment was 23 (range, 1-101) and 35 months (range, 4-101), respectively, for the whole cohort and first-line IVIg (supplemental Figure 3B-C).
Different types of therapies used in first and subsequent lines of treatment (n = 135) are summarized in Table 4. The most frequently used treatments were IVIg (n = 55/135, 41%), rituximab-based regimens (n = 19, 14%), corticosteroids (n = 14, 10%), immunosuppressive therapies (n = 14, 10%), plasma exchange (n = 8, 6%), chlorambucil (n = 6, 4%), and other chemotherapy regimens (n = 5, 4%). Although IVIg and rituximab-based regimens were associated with overall clinical responses of 53% and 52%, respectively, corticosteroids and immunosuppressive therapies were largely ineffective with 14% and 0% of overall clinical responses, respectively (Table 4; supplemental Figure 2).
Therapies used in first-line and subsequent lines of treatment and clinical responses
| All lines of treatment | n | Clinical responses, n (%) | |||
|---|---|---|---|---|---|
| CR | PR | SD | Progression | ||
| IVIg | 55 | 5 (9) | 24 (44) | 16 (29) | 10 (18) |
| IVIg and rituximab | 7 | 1 (14) | 1 (14) | 4 (58) | 1 (14) |
| Rituximab-based regimens | 19 | 3 (16) | 7 (36) | 6 (32) | 3 (16) |
| Monotherapy | 13 | 1 (8) | 6 (46) | 4 (31) | 2 (15) |
| Rituximab, cyclophosphamide | 2 | 0 (0) | 1 (50) | 0 (0) | 1 (50) |
| Rituximab, fludarabine | 2 | 1 (50) | 0 (0) | 1 (50) | 0 (0) |
| Rituximab, corticoids | 1 | 1 (100) | 0 (0) | 0 (0) | 0 (0) |
| Rituximab, ibrutinib | 1 | 0 (0) | 0 (0) | 1 (100) | 0 (0) |
| Corticosteroids | 15 | 0 (0) | 2 (13) | 2 (13) | 11 (74) |
| Immunosuppressive therapies | 15 | 0 (0) | 0 (0) | 4 (27) | 11 (73) |
| Azathioprine | 6 | 0 (0) | 0 (0) | 2 (33) | 4 (67) |
| Cyclophosphamide | 6 | 0 (0) | 0 (0) | 2 (33) | 4 (67) |
| Cyclosporine | 1 | 0 (0) | 0 (0) | 0 (0) | 1 (100) |
| Mycophenolate mofetil | 1 | 0 (0) | 0 (0) | 0 (0) | 1 (100) |
| Methotrexate | 1 | 0 (0) | 0 (0) | 0 (0) | 1 (100) |
| Plasma exchange | 10 | 1 (10) | 3 (30) | 0 (0) | 6 (60) |
| Chlorambucil | 9 | 0 (0) | 3 (33) | 0 (0) | 6 (67) |
| Other chemotherapy regimens | 5 | 0 (0) | 2 (67) | 1 (33) | 0 (0) |
| CHOP | 3 | 0 (0) | 1 (100) | 0 (0) | 0 (0) |
| Fludarabine, cyclophosphamide | 1 | 0 (0) | 1 (100) | 0 (0) | 0 (0) |
| Ibrutinib | 1 | 0 (0) | 0 (0) | 1 (100) | 0 (0) |
| Total | 135 | 10 (8) | 42 (32) | 33 (25) | 48 (35) |
| All lines of treatment | n | Clinical responses, n (%) | |||
|---|---|---|---|---|---|
| CR | PR | SD | Progression | ||
| IVIg | 55 | 5 (9) | 24 (44) | 16 (29) | 10 (18) |
| IVIg and rituximab | 7 | 1 (14) | 1 (14) | 4 (58) | 1 (14) |
| Rituximab-based regimens | 19 | 3 (16) | 7 (36) | 6 (32) | 3 (16) |
| Monotherapy | 13 | 1 (8) | 6 (46) | 4 (31) | 2 (15) |
| Rituximab, cyclophosphamide | 2 | 0 (0) | 1 (50) | 0 (0) | 1 (50) |
| Rituximab, fludarabine | 2 | 1 (50) | 0 (0) | 1 (50) | 0 (0) |
| Rituximab, corticoids | 1 | 1 (100) | 0 (0) | 0 (0) | 0 (0) |
| Rituximab, ibrutinib | 1 | 0 (0) | 0 (0) | 1 (100) | 0 (0) |
| Corticosteroids | 15 | 0 (0) | 2 (13) | 2 (13) | 11 (74) |
| Immunosuppressive therapies | 15 | 0 (0) | 0 (0) | 4 (27) | 11 (73) |
| Azathioprine | 6 | 0 (0) | 0 (0) | 2 (33) | 4 (67) |
| Cyclophosphamide | 6 | 0 (0) | 0 (0) | 2 (33) | 4 (67) |
| Cyclosporine | 1 | 0 (0) | 0 (0) | 0 (0) | 1 (100) |
| Mycophenolate mofetil | 1 | 0 (0) | 0 (0) | 0 (0) | 1 (100) |
| Methotrexate | 1 | 0 (0) | 0 (0) | 0 (0) | 1 (100) |
| Plasma exchange | 10 | 1 (10) | 3 (30) | 0 (0) | 6 (60) |
| Chlorambucil | 9 | 0 (0) | 3 (33) | 0 (0) | 6 (67) |
| Other chemotherapy regimens | 5 | 0 (0) | 2 (67) | 1 (33) | 0 (0) |
| CHOP | 3 | 0 (0) | 1 (100) | 0 (0) | 0 (0) |
| Fludarabine, cyclophosphamide | 1 | 0 (0) | 1 (100) | 0 (0) | 0 (0) |
| Ibrutinib | 1 | 0 (0) | 0 (0) | 1 (100) | 0 (0) |
| Total | 135 | 10 (8) | 42 (32) | 33 (25) | 48 (35) |
With a median follow-up time from the onset of neurologic symptoms of 127 months (range, 1-434 months) for the whole cohort, 30 (65%) patients were still alive at their last follow-up. The median OS of our cohort was not reached (range, 0.16-31 years) (supplemental Figure 3A). Deaths were related to CANOMAD/CANDA, hematologic malignancies, or other causes in 5 (33%), 3 (20%), and 7 (47%) cases, respectively. Others causes included progressive solid tumors (n = 3, comprising the aforementioned bronchopulmonary cancer [n = 2] and glioblastoma [n = 1]), infectious (n = 2), and cardiovascular (n = 2) complications.
Rituximab-based regimens
Nineteen patients were treated with a regimen including rituximab in first-line (n = 1) or subsequent lines of treatment (n = 18). Five patients received a rituximab-based regimen twice or more. The median line of treatment of rituximab-based regimens was 3 (range, 1-7). Tolerance of rituximab was excellent with the exception of 1 anaphylactic shock that resolved after corticosteroid therapy. No IgM flare was reported.
Rituximab-based regimens consisted of rituximab monotherapy (n = 13), immunochemotherapy combination (n = 4; rituximab-fludarabine, n = 2; rituximab-cyclophosphamide-dexamethasone, n = 2), or other regimens (rituximab-ibrutinib, n = 1; rituximab-corticosteroids, n = 1). Immunochemotherapy was used in patients with an overt hematologic malignancy (WM, n = 3; marginal zone lymphoma, n = 1). Five patients were retreated with a second course of single-agent rituximab on disease progression. This rechallenge led to similar or improved clinical responses. Biological responses to rituximab-based regimens were available or interpretable (pretreatment IgM peak value >0.5 g/L) in 14 of 19 cases, correlated well to clinical responses, and were distributed as follows: 2 CR (corresponding to 1 clinical CR and 1 clinical PR), 6 PR (corresponding to 5 clinical PR and 1 clinical PR), 5 SD (corresponding to 4 clinical SD and 1 clinical progression), and 1 progression (corresponding to 1 clinical progression; Figure 1; supplemental Table 2). Considering first use of rituximab-based regimens (n = 14), median time to next treatment was 45 months (range, 2-100 months; supplemental Figure 3D).
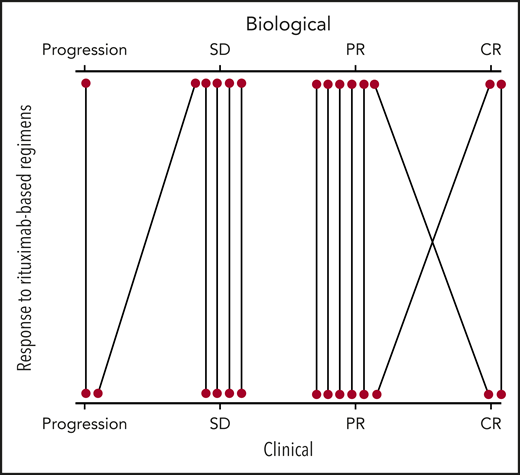
Evaluation of clinical responses (bottom) according to biological responses (top) for patients that received rituximab-based regimens. Fourteen patients were evaluable for biological response (supplemental Table 2).
Evaluation of clinical responses (bottom) according to biological responses (top) for patients that received rituximab-based regimens. Fourteen patients were evaluable for biological response (supplemental Table 2).
Five additional patients received both single-agent rituximab and IVIg that precluded single evaluation of each drug efficacy (Table 3).
Discussion
To our knowledge, this is the largest retrospective series of CANOMAD/CANDA available to date. In this series, we confirmed many of the already published clinical features, extended our knowledge of different clinical presentations, characterized the spectrum of associated hematologic malignancies, and highlighted IVIg and rituximab-based regimens efficacy.
Consistent with previous studies, most patients were male, in the fifth decade of life, and were disabled by sensory ataxia. However, even if sensory symptoms were predominant, they were not specific, and clinical presentation in our series was less homogeneous than previously described.6 Only 19 of 45 (42%) patients recapitulated all the features of CANOMAD. Ophthalmoplegia was only observed in 44%, and severe forms (modified Rankin score of 4-5) were not rare (27%). Also, in keeping with others studies, all patients had antiganglioside antibodies with IgM specificity, reacting against GD1b, GD3, GT1b, or GQ1b, and around half of the cohort had antibodies directed against these 4 disialosyl epitopes, emphasizing that they are key biological elements for diagnosis. All the data mentioned above plead for a more extensive entity that the acronym CANDA could better encompass, as previously suggested.8 From a practical point of view, CANOMAD/CANDA must be suspected in case of sensory symptoms associated with ocular or bulbar motor disorders but also in case of isolated sensory forms especially if (1) there is ataxia (present in half of cases), (2) the disease progresses in flare-ups, and/or (3) anti-MAG antibodies are negative. This also emphasizes the need for cooperation between the hematologist and neurologist in difficult cases.
This study is the first to describe comprehensively the spectrum of hematologic malignancies associated with CANOMAD/CANDA. All patients had detectable serum monoclonal IgM. Although no monotypic light chain predominance was observed, there is a possible bias toward λ expression (49%) in our cohort in comparison with the 70% to 80% of monotypic κ expression usually reported in IgM monoclonal gammopathy of unknown significance.33 As anticipated by the presence of monoclonal IgM, the identified associated hematologic malignancies were B-cell lymphoproliferative disorders, which accounted for around one third of the cohort and mainly consisted of WM (56%). Of note, only 31 (69%) patients of our cohort had bone marrow examination, possibly underestimating the proportion of patients with overt associated hematologic malignancy.
Clinical features of CANOMAD/CANDA can precede, be concurrent to, or appear during follow-up of a previously treated hematologic malignancy. The clinical course was highly variable, with some patients being wheelchair users after a few months despite therapeutic intervention and others remaining mobile after more than 10 years of follow-up without any treatment. Treatments used in our CANOMAD/CANDA series were very heterogeneous (>10), but most of them corresponded to those used in chronic inflammatory demyelinating polyneuropathy (CIDP). Indeed, as for CIDP, IVIg and corticosteroids (31/41, 75%) represented the most frequent first-line treatments. Global overall clinical responses to first-line treatment in our cohort was 44% and was mostly seen in patients treated with IVIg (60%). Efficacy rate of IVIg was in accordance with those reported in previous studies in CIDP (40% to 60%)34 and CANOMAD/CANDA (55% to 68%).7,23 Of note, the efficacy of IVIg was similar whether used as first-line treatment or in the relapse/refractory setting, with overall clinical responses obtained in around half of the cases. In contrast to its observed efficacy in CIDP (40% to 60%)34 and previous experiences in CANOMAD/CANDA (30% to 50%),7,23 corticosteroids were very rarely effective (13%) in our series. Although less frequently used, plasma exchange efficacy was also similar to previous reports, with clinical responses observed in 40% to 50% of cases.
Considering the constant presence of a monoclonal IgM that produces the antiganglioside antibody activity and recapitulates clinical features, CANOMAD/CANDA should be considered as a neurologic subtype of MGCS.1 In this regard, our data highlighted the benefit of targeting the B-cell clone in CANOMAD/CANDA as already has been done in other types of MGCS. Rituximab-based regimens showed an efficacy rate of 53%, similar to the one observed with IVIg. They were mostly used in second or subsequent lines of treatment in patients that already experimented IVIg. Efficacy of rituximab-based regimens was comparable in patients with or without an associated hematologic malignancy. These results are in line with the efficacy reported with rituximab in anti-MAG neuropathy35,36 and in non-CANOMAD/CANDA CIDP associated with hematologic malignancies.37
This study has limitations, in part, because of its retrospective design encompassing a long period of time. Data are lacking regarding delay of response to treatment, preventing us from drawing conclusions as to which treatment could be better suited to patients with rapidly progressive disease. Additionally, clinical evaluation was not centrally reviewed, although all patients were followed up in CANOMAD neurologic reference centers.
Several questions remain unanswered about the management of CANOMAD/CANDA patients. Further studies will determine whether rituximab can allow safe withdrawal of IVIg in responding but dependent patients. For patients whose response to rituximab is insufficient, immunochemotherapy with adjunction of alkylating agents may improve outcomes obtained with single-agent rituximab, as has been reported for anti-MAG neuropathy.38 Alternatively, more recently developed targeted B-cell drugs such as Bruton tyrosine kinase and Bcl-2 inhibitors, which have demonstrated clinical efficacy in treatment of WM and other B-cell lymphoproliferative disorders,39,40 could be of potential benefit.
Based on our cohort and systematic analysis of the literature, we propose a therapeutic algorithm for CANOMAD/CANDA management (Figure 2). First-line IVIg treatment appears to be the most efficient and validated therapeutic option. IVIg refractory or relapsed forms require rituximab-based regimens as a single agent or in association with alkylating agents. IVIg rechallenge could be proposed in case of long-lasting remission after first-line IVIg treatment.
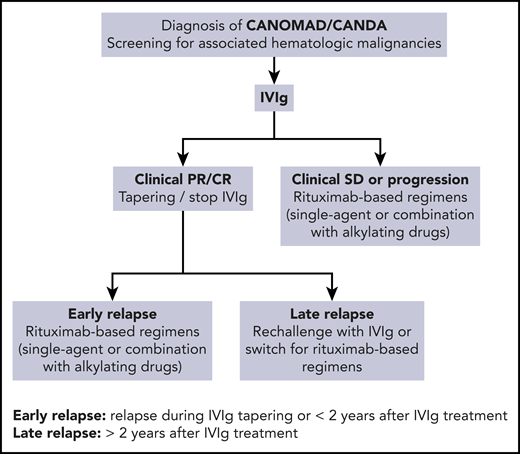
In conclusion, CANOMAD/CANDA is a rare, frequently debilitating, and probably underdiagnosed syndrome, which is associated with an overt hematologic malignancy, mainly WM, in one third of patients. IVIg and rituximab-based regimens are the most effective therapies. Corticosteroids and immunosuppressive drugs should not be used. Similar to anti-MAG neuropathy, CANOMAD/CANDA should be considered a neurologic subtype of MGCS and therefore could benefit from B-cell–directed therapies. More studies are warranted to better characterize the correlation between clinical and biological responses and define the optimal therapeutic options and sequence.
For original data, please contact the corresponding author at damien.roosweil@aphp.fr.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This study was supported (in part) by INCA-DGOS-INSERM_12560. SiRIC CURAMUS is financially supported by the French National Cancer Institute, the French Ministry of Solidarity and Health, and INSERM with financial support from ITMO Cancer AVIESAN (Alliance Nationale pour les Sciences de la Vie et de la Santé/National Alliance for Life Sciences & Health).
Authorship
Contribution: M.L.C., F.B., K.V., L.S., V.L., and D.R.-W. designed the research, analyzed data, and wrote the manuscript; M.L.C., F.B., K.V., L.S., C.T., C.R., G.M., E.L., L.M., E.D., M.M., J.F., A.E.-L., J.-C.A., M.B., B.A., A.P., A.C., T.M., V.L., and D.R.-W. recruited patients; F.B. centrally reviewed all electrophysiological data; and all authors reviewed and approved the manuscript.
Conflict-of-interest disclosure: V.L. reports receiving honoraria and advisory board fees from Roche. The remaining authors declare no competing financial interests.
Complete lists of the members of the French CIDP and FILO Groups appear in the supplemental appendix.
Correspondence: Damien Roos-Weil, Sorbonne Université, AP-HP, Hôpital Pitié-Salpêtrière, Service d’Hématologie Clinique, 47-83 Bd de l’Hôpital, 75651 Paris Cedex, France; e-mail: damien.roosweil@aphp.fr.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal