

In this issue of Blood,1 investigate genome-wide chromosomal copy gains and losses in breast impant–associated anaplastic large cell lymphomas (BIA-ALCLs) and identify that frequent losses at chromosome 20q13.13 are a characteristic genomic feature of this disease.
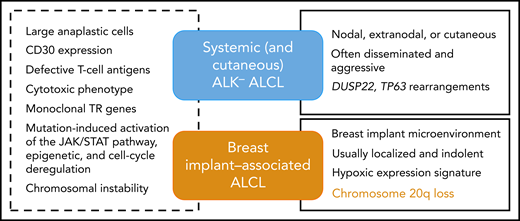
BIA-ALCL is a very rare T-cell lymphoma categorized in the current World Health Organization classification of lymphoid malignancies as a provisional entity. It is defined as a subtype of anaplastic lymphoma kinase (ALK)− anaplastic large cell lymphoma (ALCL), which arises in patients with breast implants inserted for either cosmetic or reconstructive purposes.2 The disease typically presents as a late-onset pericapsular effusion (seroma-associated or in situ lymphoma) and is usually cured by complete surgical excision. Less commonly, patients are diagnosed with poor prognosis, advanced stage disease with an infiltrative tumor mass, or with regional lymph node involvement.3 Since the first case was described in 1997, epidemiological studies have confirmed that there is a causal relationship to the presence of textured breast implants. Our current understanding of BIA-ALCL pathogenesis includes a chronic inflammatory/immune reaction elicited by the implant or bacteria adherent to it, with secondary genetic lesions mediating transformation, dependence on cytokine activation, and JAK-STAT pathway activation.3,4
Because of the rarity of the disease and usually limited availability of tumor cells or tissues, only a small number of cases have been evaluated by genomic and molecular techniques. Previous studies have focused on characterizing the mutational landscape of BIA-ALCL or on searching for structural variants known to occur in other ALK− ALCLs.5 In the paper published in this issue, the authors used shallow (low-coverage) whole-genome sequencing in order to evaluate chromosomal copy number aberrations in BIA-ALCL and compare the results to systemic ALK− ALCL. Shallow whole-genome sequencing is a recently described technique of copy number variation sequencing, which requires a depth of genome coverage <1 time. It uses shortread sequencing, making it particularly suitable for analyzing DNA from routinely processed formalin-fixed samples and is based on the counts of reads aligned to chromosomal windows or bins after exclusion of problematic genomic regions.6 Limitations of the method include that it does not identify structural chromosomal aberrations or polyploidy. Genome-wide DNA profiling of BIA- and systemic ALCLs detected a broad range of genomic imbalances in the vast majority of cases, both gains and losses, distributed along all chromosomes, indicating that chromosomal instability already documented in systemic ALCLs is a shared feature of both entities. This is also in line with cytogenetic data from the few BIA-ALCL cell lines consistently showing complex karyotypes.7 There was substantial overlap and few significant differences between the 2 entities, but strikingly the most frequent aberration in BIA-ALCL (ie, 20q loss peaking at 20q13.13-13.2 present in two-thirds of the cases) was detected in only 13% of the systemic ALK− ALCLs analyzed. Moreover, 20q loss has been very rarely found in other ALCLs and peripheral T-cell lymphoma-not otherwise specified. Therefore, this finding qualifies 20q deletion as a genomic marker unique to BIA-ALCL. This feature is important for disease definition and classification because it supports the concept that this a separate entity distinct from other ALK− ALCLs (see figure). Indeed, besides the unique clinical context being 1 major defining feature of the disease, BIA-ALCL is otherwise indistinguishable from other ALK− ALCLs by morphology and immunophenotype. Moreover, the pattern of mutations observed in BIA-ALCL, preferentially distributed among effector and regulators of the JAK/STAT pathway, epigenetic modifiers, and regulators of cell cycle is overlapping with that of other T-cell lymphoma entities, especially other ALK− ALCLs.4 Of note, in line with the novel findings reported here, 2 studies have also documented specificities in terms of the gene expression signature of BIA-ALCL, notable for an hypoxic signature, which likely reflects the peculiar confined microenvironment in which this lymphoma develops.8,9
An interesting finding reported by Los-de Vries et al is that in situ (seroma-type) BIA-ALCL comprises a more aberrant and more heterogeneous genome compared with invasive tumors, whereas an inverse relationship was described for mutations, more numerous in tumor-type BIA-ALCL.4 This brings a meaningful insight into BIA-ALCL progression, which seems to be associated with a change in oncogenic signatures10 and a selection of subclones.
Whether shallow whole-genome profiling represents a useful adjunct in the assessment of periprosthetic seroma fluid requires prospective evaluation. Feasibility can be inferred from the conclusive results on circulating free DNA, but the major obstacle lies in the availability of the technique. It is not yet widely used in diagnostic laboratories. A systematic analysis of seroma fluids, assuming sufficient samples can be obtained, would contribute interesting insights into currently poorly characterized early stages of BIA-ALCL development.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal