

Local inflammation is characterized by leukocytes that migrate out of the circulation by penetrating the vascular endothelial lining. It is remarkable that during this event, there is hardly any leakage. How does the endothelium manage to maintain its integrity while numerous leukocytes penetrate? That is one of the key questions in the field of inflammation, and in this issue of Blood, may have provided the answer.1
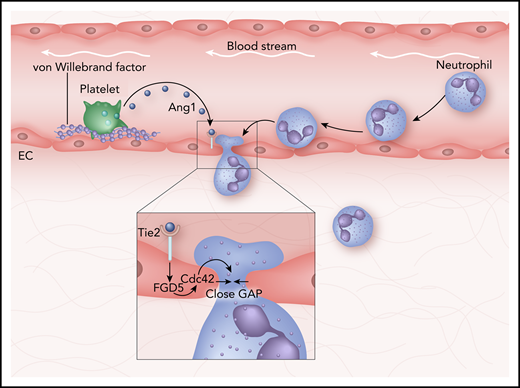
How the vascular gap is closed after a neutrophil has crossed. A neutrophil leaves the circulation by penetrating in between two individual endothelial cells (ECs). Consequently, it leaves behind a gap in the endothelial lining. Platelets stick to von Willebrand factor, release angiopoeitin-1 (Ang1), resulting in activating the endothelial receptor Tie2. Inset: Tie2 activation triggers intracellular endothelial signals, including the actin remodelling proteins RhoGEF FGD5 and small RhoGTPase Cdc42, resulting in lateral protrusions and closing of the gap.
How the vascular gap is closed after a neutrophil has crossed. A neutrophil leaves the circulation by penetrating in between two individual endothelial cells (ECs). Consequently, it leaves behind a gap in the endothelial lining. Platelets stick to von Willebrand factor, release angiopoeitin-1 (Ang1), resulting in activating the endothelial receptor Tie2. Inset: Tie2 activation triggers intracellular endothelial signals, including the actin remodelling proteins RhoGEF FGD5 and small RhoGTPase Cdc42, resulting in lateral protrusions and closing of the gap.
The authors show that platelets are essential for closing the endothelial gaps that are induced by the transmigrating neutrophils. This dynamic interplay between platelets and the endothelium depends on endothelial-derived von Willebrand factor acting as a glue for the platelets to bind to the endothelium. As a result, platelets release angiopoietin-1, which then binds to its receptor Tie-2 on the endothelium. This triggers an intracellular response from the endothelium consisting of actin remodeling and lateral protrusions that close the gap once the neutrophil has crossed (see figure).
For years, researchers argued that when leukocytes cross the vessel wall under normal inflammatory conditions, permeability is simultaneously increased. However, elegant work from the laboratory of Baluk et al2 in the 1990s using electron microscopy on in vivo lung tissues showed that the locations where these 2 events take place differ. By using biotinylated lectins or silver nitrate to stain endothelial cells in situ and Monastral blue as a tracer to quantify plasma leakage, those studies showed that most of the leakage occurred in post capillary venules (<40 μm diameter), whereas most of the leukocyte migration (predominantly neutrophils) occurred in collecting venules, with the notion that capillaries and arterioles did not leak. Additional studies complemented those findings by showing that vascular permeability required actomyosin-related tension, whereas neutrophil transmigration was independent of such triggers.3 Moreover, permeability-inducing factors such as histamine along with VEGF and leukocyte transmigration could trigger the phosphorylation levels of the endothelial cell-cell junction molecule vascular endothelial cadherin (VE-cadherin).4 But those events induced the phosphorylation of different tyrosine residues on VE-cadherin, suggesting that both processes mediated the opening of endothelial junctions in different ways.
The outstanding question is How does the endothelium close the gap, once a leukocyte has passed? Heemskerk et al identified the mechanism responsible for this, and it includes local activation of RhoA, a small RhoGTPase.5 They showed that the endothelium, in response to the transmigrating leukocyte, induces a RhoA-mediated F-actin–rich ring around a leukocyte once it transmigrates through the endothelial barrier.5 This actin ring functions as an elastic strap that tightly seals the endothelial membrane around the transmigrating leukocyte and thereby limits local vascular leakage during transmigration. Although ICAM-1 signaling was implicated in this pathway as the initial upstream trigger, it became clear that it was not the only upstream activator involved in closing the gap.
Evidence that the endothelium itself is involved in restoring the gaps after leukocytes have crossed came from the realization that endothelial cells can induce membrane protrusions around transmigrating leukocytes.6,7 To this day, the function of these protrusions remains elusive. It was proposed that such protrusions are involved in the adhesion step6 or may be important for the actual diapedesis step.7 Alternatively, it was hypothesized that these protrusions could potentially close the gaps, induced by the transmigrating neutrophil.8 No clear function can be attributed to these protrusions yet, but it is evident that they are involved in the transmigration process.
Interestingly, it was found that platelet depletion led to inflammatory bleedings, independent of coagulation-related mechanisms.9 A potential mechanism has been proposed in which platelets would plug vascular gaps to prevent leakage.10 Alternatively, factors released by platelets may initiate signals in the endothelium that would trigger gap sealing. In support of this latter idea, platelet granules contain several factors that can stabilize endothelial cell-cell junctions.11
For endothelial cells to be able to close the gap when neutrophils cross, the extracellular signals must originate from a local source. Braun et al show that angiopoietin-1 is released from platelets and acts on the endothelial Tie-2 receptor, triggering the RhoGEF FGD5 to activate the small RhoGTPase Cdc42 (see figure). This leads to the local induction of lateral protrusions that effectively seal the gap. For this to happen, platelets need to be locally recruited to the sites where neutrophils exit the endothelium. Indeed, Braun et al found that platelets localized at sites where neutrophils exit the vasculature. How platelets are actively recruited to be in close proximity with such neutrophil exit sites remains unclear. Although it has been recognized that leukocytes can form complexes with circulating platelets,12 it will be interesting to investigate the regulatory role of these complexes for efficient inflammation-related and leakage-preventing transmigration events.
Physiologically, it is important that the endothelial gap is efficiently and rapidly closed upon leukocyte exiting. If it is not, this can lead to severe pathological defects in remote organs, most notably in the lungs.13 Once more, this work shows us the importance of understanding basic mechanisms so we can develop effective therapies to target vascular leakage syndromes during pathophysiological inflammatory conditions.
Conflict-of-interest disclosure: The author declares no competing financial interests.
