

In this issue of Blood, report that hepatic transferrin (Trf) has a pivotal role in the regulation of systematic iron metabolism, erythropoiesis, and in the protection of liver from iron overload-evoked ferroptosis, fibrosis, and cirrhosis.1 Based on their novel findings, the authors propose a potential therapeutic strategy for mitigating a spectrum of iron overload disorders by targeting iron-evoked ferroptosis and/or the divalent metal transporter Slc39a14.
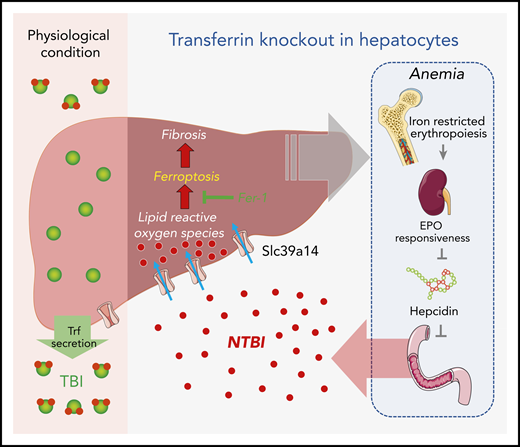
Under physiological conditions, Trf binds to ferric ion and delivers it to a variety of tissues (eg, the liver) for iron requisition and use. When hepatic Trf is absent, affected individuals develop microcytic anemia because of iron-restricted erythropoiesis, which leads to increased iron absorption in the intestine as a consequence of erythropoietin/erythroferrone-dependent suppression of hepcidin, NTBI accumulation via Slc39a14 in multiple organs, and finally the iron overload–evoked ferroptosis, fibrosis, and cirrhosis. EPO, erythropoietin.
Under physiological conditions, Trf binds to ferric ion and delivers it to a variety of tissues (eg, the liver) for iron requisition and use. When hepatic Trf is absent, affected individuals develop microcytic anemia because of iron-restricted erythropoiesis, which leads to increased iron absorption in the intestine as a consequence of erythropoietin/erythroferrone-dependent suppression of hepcidin, NTBI accumulation via Slc39a14 in multiple organs, and finally the iron overload–evoked ferroptosis, fibrosis, and cirrhosis. EPO, erythropoietin.
It is well known that the trace element iron is required for the normal function of many tissues and that abnormalities of iron metabolism contribute to disease in humans, but the underlying mechanisms remain incompletely understood. Trf, encoded by the Trf gene, is a blood plasma glycoprotein that controls free iron (Fe) levels by binding to ferric ion and delivering it to a variety of tissues, including the liver, bone marrow, and spleen. Trf was first identified in 1947.2 Since then, 2 of the enduring enigmas in the field of iron biology are Which tissues produce Trf? and What is the role of tissue-specific Trf? Historically, much of our knowledge of Trf was derived from studies of atransferrinemia, a rare autosomal recessive metabolic disorder that reduces Trf to undetectable levels and causes severe anemia, liver iron overload, and fibrosis.3 More recently, much has been learned from the hypotransferrinemic (Hpx) murine model of atransferrinemia, attained by a point mutation in the Trf gene yielding less than 1% of the normal Trf expression. Hpx mice cannot survive unless exogenous Trf is supplemented during the first 2 weeks after birth.3,4 Trf mutations have been shown to attenuate iron binding at either the N lobe and/or C lobe of Trf in mice. Affected mutants display hepatocellular iron overload with decreased hepcidin expression.5 However, the liver-specific role of Trf has not been described in detail.
To gain insight into the role of hepatic Trf in iron metabolism, Yu et al generated and characterized the hepatocyte-specific Trf knockout mice, Trf-LKO. The Trf-LKO mutants developed hypoferric anemia as a result of iron-restricted erythropoiesis, which was associated with dampened erythropoietin responsiveness, elevated erythroferrone expression, and dysregulation of iron metabolism. There was a marked reduction of Trf-bound iron (TBI) in serum and an overload of non–Trf-binding iron (NTBI) in various tissues, particularly the liver. The hepatic pathology worsened with elevated dietary iron absorption and subsequent tissue iron accumulation as the consequence of hepcidin suppression by the erythropoietin-dependent induction of erythroferrone (see figure). In contrast to the well-studied Hpx mice, this liver-specific Trf knockout mouse model is genetically and phenotypically distinct. Notably, the Trf-LKO mouse model allows for the study of Trf in a tissue-specific manner, in particular its intra- vs extra-hepatic functions. The findings from this study indicate that hepatic Trf is indispensable for the maintenance of iron homeostasis and hematopoiesis and that extra-hepatic Trf can partially compensate for the loss of hepatic Trf. Conceivably, further exploration of the extra-hepatic Trf function using these genetically modified animals would be of great interest.
Insights from this study greatly inform our understanding of the role of Trf in the pathology of iron overload-evoked hepatic injury, and other iron-related organ disorders. The liver, as the primary Trf-producing organ, is crucial to iron metabolism and homeostasis. However, whether hepatic Trf has a relationship with liver fibrosis was formerly unknown. Using multiple linear regression analysis, Yu et al found that patients with liver fibrosis had significantly reduced serum Trf, which was inversely correlated with the level of hepatic fibrosis markers. These findings were reproduced in animal model studies. Mice with hepatic Trf deletion, when fed with a high iron diet or subjected to CCl4-induced liver injury had elevated NTBI accumulation, enhanced liver ferroptosis, and subsequent fibrosis. These findings are consistent with and complementary to the work published previously by this team and others, demonstrating that ferroptosis contributes to a spectrum of disorders, including neurodegeneration6 , cardiomyopathy,7 and hemochromatosis.8 These exciting and important findings collectively provide the rationale for attributing iron metabolic deregulation and ferroptosis in the etiology of certain human diseases and in developing novel therapeutics that correct iron metabolic disorders.
After demonstrating the toxicity of excessive NTBI in the liver, Yu et al explored how NTBI enters various types of cells, particularly hepatocytes. Slc39a14 is a transmembrane-bound metal transporter primarily responsible for the cellular uptake of manganese and zinc under physiological conditions. In their study, the authors uncovered an important role of Slc39a14 in transferring NTBI into hepatocytes when hepatic Trf levels were reduced, which resolved a long-standing myth about Hpx mice.3 Notably, the liver-specific deletion of Slc39a14 was found to partially inhibit cell ferroptosis and to protect the liver from fibrosis in the animal models (see figure). This finding was consistent with data from the article’s clinical studies, which found that patients with liver cirrhosis have reduced hepatic Trf, elevated Slc39a14 expression, and increased iron deposition. Taken together, these data provide strong evidence that Slc39a14 participates in the active transportation of NTBI into cells—especially hepatocytes—and that hepatic Slc39a14 could be a potential therapeutic target for preventing and treating ferroptosis-evoked liver damage.
The novel findings of this study are important and of great translational potential. Liver fibrosis is one of the most common non-cancer causes of death worldwide, and the search for novel therapeutics is ongoing. In their study, Yu et al convincingly demonstrate that iron overload–evoked ferroptosis participates in liver fibrosis and cirrhosis in both the animal models and in human clinical disease. Moreover, inhibition of ferroptosis by ferrostatin-1, the attenuation of iron overload by the dietary supplementation of apo-Trf, or the deletion of Slc39a14 in mice can significantly alleviate excessive serum NTBI, liver iron deposition, and liver fibrosis. The work described by Yu et al builds a strong foundation on which novel therapeutics for liver fibrosis could be developed by inhibiting iron-evoked ferroptosis and by preventing hepatic iron overload. Moreover, a thorough understanding of the modulation of iron metabolism and homeostasis is instrumental to the establishment of dietary guidelines and to the mitigation of other iron overload (eg, hereditary hemochromatosis) and deficient (eg, as iron deficiency anemia) diseases.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal