

In this issue of Blood, 1 used a cell-based screening to identify 9 unrelated drugs that may cause bleeding by interfering with the vitamin K (VK) cycle that is required for the VK-dependent γ-carboxylation of blood coagulation proteins. Bleeding caused by drugs inhibiting the reduction of VK epoxide, but not VK, can be rescued by VK administration.
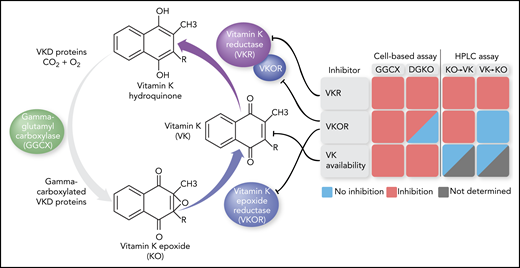
The VK cycle in which GGCX adds a carboxylic acid to specific Glu residues in VKD proteins, thereby using VK hydroquinone as cosubstrate. VKOR reduces the generated KO to VK, which is further reduced by VKOR and VKR. Monitoring reporter protein γ-carboxylation in GGCX or VKOR (DGKO) knockout cell lines using KO as substrate and quantifying KO/VK formation via high-performance liquid chromatography (HPLC) analysis led to the identification of drugs that inhibited VKOR, VKR, or intracellular VK availability. With regard to the last, small amounts of intracellular VK precluded accurate KO/VK assessment for 1 drug (“Not determined”). The figure has been adapted from Figure 1A in the article by Chen et al that begins on page 898.
The VK cycle in which GGCX adds a carboxylic acid to specific Glu residues in VKD proteins, thereby using VK hydroquinone as cosubstrate. VKOR reduces the generated KO to VK, which is further reduced by VKOR and VKR. Monitoring reporter protein γ-carboxylation in GGCX or VKOR (DGKO) knockout cell lines using KO as substrate and quantifying KO/VK formation via high-performance liquid chromatography (HPLC) analysis led to the identification of drugs that inhibited VKOR, VKR, or intracellular VK availability. With regard to the last, small amounts of intracellular VK precluded accurate KO/VK assessment for 1 drug (“Not determined”). The figure has been adapted from Figure 1A in the article by Chen et al that begins on page 898.
Adverse drug reactions, and, in particular, drug-induced bleeding, are among the leading causes of hospitalization and death. Bleeding is associated with the use of anticoagulants, as well as with nonsteroidal anti-inflammatory agents, serotonin reuptake inhibitors, and antibiotics. With regard to the last, the β-lactam antibiotic subgroup cephalosporins that contain an N-methylthiotetrazole side chain are well known to interfere with the VK cycle.2 In this cycle, VK is reduced to VK hydroquinone by VK epoxide reductase (VKOR) and an unidentified VK reductase (VKR) (see figure).3 VK hydroquinone serves as a cosubstrate for γ-glutamyl carboxylase (GGCX) that adds a carboxylic acid to specific Glu residues in VK-dependent (VKD) blood-coagulation proteins, an essential step to attain full clotting functionality. The generated VK epoxide (KO) is recycled back to VK by VKOR.
Previously, Chen et al were able to study the VK cycle in more detail by developing an elegant cell-based system that allowed for functional assessment of its components in the cell milieu. Using genome editing to knock out endogenous GGCX4 or both VKOR and a VKOR-like homolog5 and introducing mutant patient variants of these enzymes in HEK293 cells, they explained the clinical manifestations of specific mutations that included a VK-responsive bleeding phenotype and sensitivity to the VK antagonist warfarin. Taking advantage of the developed technology to identify VK cycle–targeting drugs, Chen and colleagues now performed high-throughput screening of a drug library in the GGCX-knockout cell line using KO as substrate. Twenty-two drugs were uncovered that impacted the biosynthesis of the VKD reporter protein, 9 of which appeared to be true inhibitors and were not associated with cytotoxic effects on the cells. Subsequent inhibitory efficacy assessment and high-performance liquid chromatography analysis to quantify VK and KO, using a clever combination of knockout cell lines and substrates, allowed for further dissection of the pathways involved (see figure). One set of drugs, including warfarin, was shown to inhibit VKOR, whereas another set of drugs inhibited VKR, and a third set affected cellular VK availability. The anticoagulant effect of drugs targeting VKOR could be completely restored by the administration of VK in vitro and in vivo, confirming that VKOR is primarily responsible for the reduction of KO to VK. On the other hand, drugs targeting VKR could only partially be rescued by VK.
Although the observations of Chen and colleagues clearly uncover direct inhibitory actions of specific drugs on enzymes of the VK cycle, some questions are raised with regard to the clinical interpretation of these findings. From the 9 drugs identified, nitazoxanide and lansoprazole (VKOR inhibitors) and clofazimine (VKR inhibitor) demonstrated the strongest inhibitory potency toward KO-dependent γ-carboxylation of the reporter protein, apart from warfarin. However, in clinical practice, these drugs lead to sporadic bleeding only. This apparent discrepancy may result from differences in circulatory drug levels vs inhibitory potency, in addition to the in vivo drug distribution and uptake by hepatocytes. Of note, the β-lactam antibiotics (ie, cephalosporins) that are known to inhibit γ-carboxylation of VKD proteins were not identified in this study. Whether they were not part of the drug library, and, therefore, were absent from screening, is unclear, because no detailed information on the library is provided. Screening of these compounds would serve to further validate the cell-based assay.
Most of the VK cycle–targeting drugs identified here are known to interfere with VK antagonist therapy to some extent. Until now, this effect was primarily ascribed to inhibition of the hepatic cytochrome (CYP)2C9 and/or CYP3A4 enzyme systems, thereby impairing VK antagonist metabolism.6 Whether direct inhibition of the VK cycle, resulting in an impaired production of VKD proteins, contributes in a synergistic manner to the interference with VK treatment remains to be determined. Additional drug candidates for screening are the serotonin reuptake inhibitors that, although considered to primarily give rise to bleeding complications by inhibiting platelet aggregation, also inhibit CYP2C97 and, to a minor extent, CYP3A4.8 Although concomitant use of serotonin reuptake inhibitors and VK antagonists has been associated with high international normalized ratio values,9 bleeding observed in patients using both types of these drugs was found to be unrelated to CYP2C9 inhibition.10 The cell-based assay system described by Chen et al can be used to further unravel the molecular mechanisms of these drugs that are known to interfere with VK antagonist therapy but lack a well-described mode of action. Moreover, it could serve as a high-throughput platform to screen novel drugs for drug-induced bleeding resulting from a clinically relevant interaction with the VK cycle.
Conflict-of-interest disclosure: M.H.A.B. has received research funding from uniQure Biopharma B.V. F.J.M.v.d.M. has received funding for HemoNED (Dutch Registry of Patients with Hemophilia and associated Disorders) from CSL Behring, Pfizer, Bayer, Novo Nordisk, Sobi, Roche, and Octapharma.
