

Background: Myelofibrosis (MF) is a myeloproliferative neoplasm (MPN) characterized by uncontrolled inflammation and fibrotic deposition in the extracellular bone marrow space, resulting in cytopenias, constitutional symptoms, and splenomegaly. A minority of patients are eligible for allogeneic hematopoietic stem cell transplant (allo-HSCT), which can be curative but is associated with substantial risks. Janus-associated kinase inhibitors (JAKi), including ruxolitinib (Rux), are approved for treatment of MF but do not reliably alter the disease course or generate durable responses. The limited treatment options for MF after Rux failure highlight a clear unmet need.
In murine models of MPN, bromodomain and extra-terminal family protein inhibitors (BETi) reduced inflammatory cytokine levels and, combined with JAKi, reduced MF disease burden (Kleppe et al, 2018). BETi also modulated key nodes in the intrinsic apoptosis pathway and synergized with the B-cell lymphoma-2 (BCL-2) family inhibitor navitoclax (Nav) in solid tumor models (data on file).
Pan-BETi have shown activity in patients with MF, including reduction in spleen volume and improvements in symptom burden, anemia, and bone marrow fibrosis as monotherapy and in combination with Rux (Mascarenhas et al, 2019). Selective BETi may reduce off-target toxicity relative to pan-BETi.
The studies described here aim to evaluate the safety, pharmacokinetics (PK), and preliminary efficacy of ≥2 dosing regimens of 2 novel BETi: mivebresib, an oral pan-BETi that demonstrated antitumor activity in preclinical models of malignancy, and ABBV-744, a novel, potent small molecule that selectively inhibits bromodomain II of the BET family. Both BETi will be investigated as monotherapy and in combination with Nav or Rux in patients with MF.
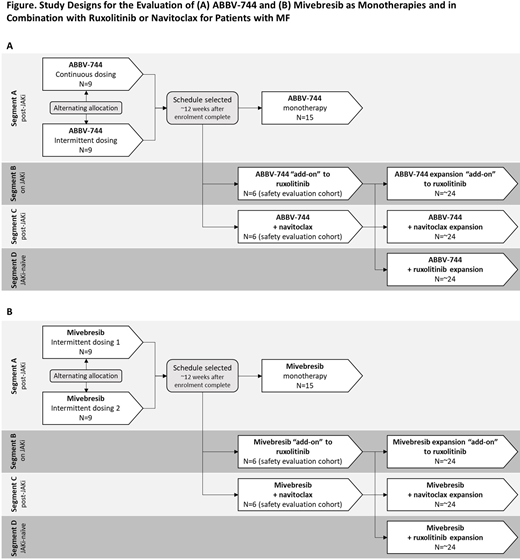
Methods: The 2 Phase 1b, multicenter, open-label studies will recruit patients with MF to receive ABBV-744 (NCT04454658) or mivebresib (NCT04480086), respectively, as monotherapy or in combination with Nav or Rux. Patients ≥18 years with intermediate-2 or high-risk MF, measurable splenomegaly (intermediate-1 with palpable splenomegaly ≥5 cm below costal margin eligible for Segment A) and Eastern Cooperative Oncology Group Performance Status <2 who are not candidates for allo-HSCT will be enrolled into 4 segments (Figure). Segment A: JAKi-experienced and BETi-naïve patients will receive 1 of 2 dosing regimens of ABBV-744 or 1 of 2 dosing regimens of mivebresib. Segment B: BETi-naïve patients currently receiving Rux will receive ABBV-744 or mivebresib plus Rux; Segment C: JAKi-experienced, and BETi-naïve, and/or BCL-XL/BCL-2 inhibitor-naïve patients will receive ABBV-744 or mivebresib plus Nav; Segment D: JAKi-naïve and BETi-naïve patients will receive ABBV-744 or mivebresib plus Rux. A dose regimen will be stopped if ≥3 of the first 9 dose-limiting toxicity (DLT)-evaluable patients given that dose in Segment A experience a DLT, or if the DLT rate is ≥33% at any point thereafter. Segments B and C will begin only once a dose regimen is deemed safe in Segment A. Segment D will commence after safety is established in Segment B. Treatment may continue until ≥1 discontinuation criterion has been met.
The primary endpoint in both studies is adverse events (AEs) defined by the Common Terminology Criteria for AEs v5, including DLTs at a dose considered tolerable in patients with MF. Key secondary endpoints include the proportion of patients with spleen volume reduction of ≥35% at Weeks 12 and 24, PK parameters, proportion of patients with ≥50% reduction in Total Symptom Score at Week 24, and objective response rate (complete remission + partial remission). Key exploratory endpoints include change from baseline in allelic frequency, improvement in bone marrow fibrosis grade, and cytokine modulation.
The planned sample size is approximately 130 patients per study. Analyses will include all patients who received ≥1 dose of study drug. Safety will be assessed by study drug exposure, AEs (including DLTs), serious AEs, deaths, and changes from baseline in laboratory determinations and vital sign parameters. AEs will include treatment-emergent events with an onset after first dose and ≤30 days after the last dose of study drug. Efficacy analyses will include summary statistics for categoric and continuous variables. Confidence intervals will be derived from the Clopper Pearson method. First dosing is planned for Q4 2020.
Mascarenhas:Incyte, Kartos, Roche, Promedior, Merck, Merus, Arog, CTI Biopharma, Janssen, and PharmaEssentia: Other: Research funding (institution); Celgene, Prelude, Galecto, Promedior, Geron, Constellation, and Incyte: Consultancy. Saab:AbbVie: Current Employment, Other: may own stock or stock options. Brackman:AbbVie Inc.: Current Employment, Other: may hold stock or other options. Modi:AbbVie: Current Employment, Other: may own stock or stock options. Abraham:AbbVie: Current Employment, Other: may own stock or stock options. Ward:AbbVie: Current Employment, Other: may own stock or stock options. Verstovsek:Celgene: Consultancy, Research Funding; Sierra Oncology: Consultancy, Research Funding; Novartis: Consultancy, Research Funding; Blueprint Medicines Corp: Research Funding; NS Pharma: Research Funding; Gilead: Research Funding; Incyte Corporation: Consultancy, Research Funding; Roche: Research Funding; Protagonist Therapeutics: Research Funding; PharmaEssentia: Research Funding; AstraZeneca: Research Funding; Genentech: Research Funding; ItalPharma: Research Funding; CTI Biopharma Corp: Research Funding; Promedior: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal