


Abstract
The last decades have seen great progress in the treatment of cold agglutinin disease (CAD). Comparative trials are lacking, and recommendations must be based mainly on nonrandomized trials and will be influenced by personal experience. Herein, current treatment options are reviewed and linked to 3 cases, each addressing specific aspects of therapy. Two major steps in CAD pathogenesis are identified, clonal B-cell lymphoproliferation and complement-mediated hemolysis, each of which constitutes a target of therapy. Although drug treatment is not always indicated, patients with symptomatic anemia or other bothersome symptoms should be treated. The importance of avoiding ineffective therapies is underscored. Corticosteroids should not be used to treat CAD. Studies on safety and efficacy of relevant drugs and combinations are briefly described. The author recommends that B cell–directed approaches remain the first choice in most patients requiring treatment. The 4-cycle bendamustine plus rituximab combination is highly efficacious and sufficiently safe and induces durable responses in most patients, but the time to response can be many months. Rituximab monotherapy should be preferred in frail patients. The complement C1s inhibitor sutimlimab is an emerging option in the second line and may also find its place in the first line in specific situations.
Introduction
Cold agglutinin disease (CAD) has a prevalence of 5 to 20 cases per million and an incidence of 0.5 to 1.9 cases per million per year, showing considerable variation with climate.1 CAD accounts for 15% to 30% of autoimmune hemolytic anemias (AIHAs).2 The author considers CAD to be a well-defined clinicopathologic entity, and the distinction between CAD and cold agglutinin syndrome (CAS) is increasingly accepted.3-6 According to recent international consensus, CAD is defined as “an AIHA with a monospecific direct antiglobulin test (DAT) strongly positive for C3d (and negative or weakly positive with IgG [immunoglobulin G]) and a cold agglutinin (CA) titer of 64 or greater at 4°C. We recognize that there may be occasional cases with CA titer <64. Patients may have a B-cell clonal lymphoproliferative disorder (LPD) detectable in blood or marrow but no clinical or radiological evidence of malignancy.”4 In contrast, CAS is a similar but heterogeneous cold hemolytic syndrome occurring secondary to another clinical disease.3,4 Only CAD will be addressed in this review.
The histopathologic substrate for CAD is indolent LPD of the bone marrow in most if not all cases.6-9 This LPD was previously perceived as being heterogeneous.10-12 In most cases, however, the bone marrow displays a characteristic histomorphologic, immune phenotypic, and molecular pattern that has been designated CA-associated LPD.6 LPD in CAD shows some common features with lymphoplasmacytic lymphoma (LPL) and marginal zone lymphoma,1,6 but the MYD88 L265P somatic mutation, present in most cases of LPL, is usually not found in CAD.6,13-15 Differences between Waldenström macroglobulinemia and CAD have also been demonstrated in immunoglobulin heavy and light chain gene use.16,17 Furthermore, typical recurrent somatic mutations and cytogenetic abnormalities have been discovered in the bone marrow of patients with CAD.8,15,18,19
CAs in CAD are monoclonal antibodies produced by clonal B cells, most often of the IgMκ class and with anti-I specificity.1,20,21 Binding of such CAs to the erythrocyte surface results in agglutination and induces complement-dependent hemolysis by activation of the classical pathway.22-25 The hemolysis is mainly extravascular, mediated by opsonization with complement protein C3b and subsequent phagocytosis, but terminal complement activation with intravascular hemolysis also occurs in some patients and situations.21-23,25,26
In a recent multinational study of 232 patients with CAD, 27% were found to have severe anemia, defined as hemoglobin (Hb) <8.0 g/dL.1,23 Anemia was moderate (Hb 8.0-10.0 g/dL) in 37% and mild (Hb 10.0 g/dL to lower limit of normal) in 24%, whereas 12% had compensated hemolysis (Figure 1). At least in cool climates, a majority of patients also have ischemic symptoms, such as acrocyanosis or Raynaud-like phenomena, resulting from agglutination of erythrocytes in the acral circulation. Complement-driven exacerbation of hemolysis is common during febrile infections and other conditions with acute-phase reaction.1,10,27,28 Estimates on transfusion dependency show considerable variation; unselected series indicate that ∼40% of patients have been transfused.1,10,11 Fatigue is a well-known symptom of unknown frequency and can be attributed to complement activation by itself as well as anemia.23,29
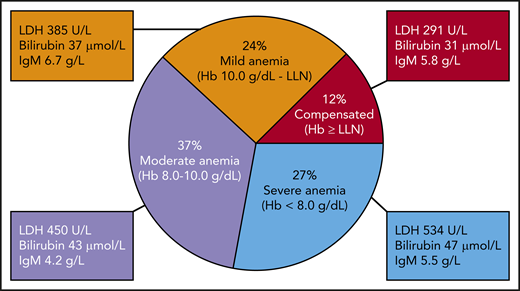
Severity of anemia in 232 patients with cold agglutinin disease. Hb levels correlate with mean values of lactate dehydrogenase (LDH) and bilirubin but not with IgM. LLN, lower limit of normal. Based on data from Berentsen et al.1
Severity of anemia in 232 patients with cold agglutinin disease. Hb levels correlate with mean values of lactate dehydrogenase (LDH) and bilirubin but not with IgM. LLN, lower limit of normal. Based on data from Berentsen et al.1
The last 2 decades have seen great progress in the development of new treatment options based on prospective trials.23,24 Each of the 2 major steps in the pathogenesis of CAD, clonal B-cell lymphoproliferation and complement-mediated hemolysis, constitutes a logical and promising target of therapy.6,24
Case 1
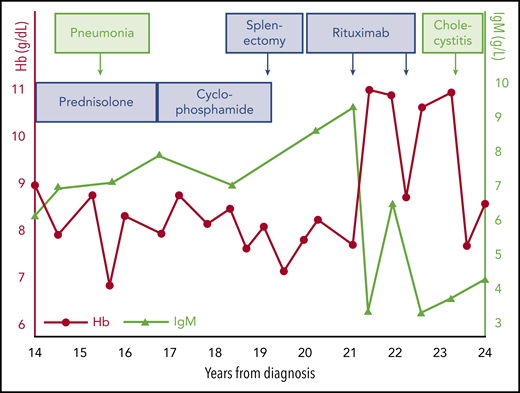
A woman participated a clinical trial at age 68 years.30 At that time, she had a 20-year history of CAD. She was unable to go out in winter or take food from the freezer or even the refrigerator without wearing gloves. At diagnosis, her Hb was 9.4 g/dL, and she had acrocyanosis. Her serum LDH level was 375 U/L (converted into today’s units), total bilirubin was 39 μmol/L, haptoglobin was not detectable, and reticulocyte count was not recorded. DAT was positive for C3d and negative for IgG. Her CA titer was 16 384 at 4°C. Some years after diagnosis, she was found to have monoclonal IgMκ with a total IgM of 6.2 g/L, and a bone marrow biopsy showed 20% lymphoid infiltration, interpreted as immunocytoma (by Kiel classification; current World Health Organization classification term, LPL).31 She had received a course of prednisolone without any effect and had then been observed untreated for 13 years, after which she was readmitted to a hematology department. Figure 2 illustrates her further history. Subsequent treatments with prednisolone and oral cyclophosphamide, respectively, had no effect but were still continued for >2 years each. She then underwent splenectomy, which also failed to improve the hemolytic anemia. Rituximab administered within a clinical trial was the first treatment to produce a meaningful response, lasting for ∼1 year. A second series of rituximab infusions was followed by a new, short-lived remission. During the course of her disease, she experienced pneumonia and acute cholecystitis, both of which were followed by a marked drop in Hb level. After having lived with CAD for 24 years, she had a stroke and died.
Diagnostic workup
The recommended diagnostic workup does not differ much from that described in case 1.4,32,33 The importance of a targeted history and clinical examination should be emphasized; this will identify the frequently occurring clinical features and contribute to the exclusion of clinically overt lymphoma (secondary CAS). The diagnosis of hemolytic anemia is based on Hb level and markers of hemolysis, such as bilirubin, LDH, and haptoglobin. Absolute reticulocyte counts will usually but not always be elevated.4,34 A monospecific (extended) DAT must be performed to confirm autoimmune pathogenesis and recognize complement activation. The typical DAT pattern in CAD is a strongly positive test for C3 (usually C3d) only, but in up to 20% of patients, DAT is weakly positive for IgG as well.1,10 Because DAT can be positive for C3 only, even in IgM-mediated warm AIHA,35,36 CA titration is mandatory for diagnosis. The CA titer is typically ≥64 but usually much higher.1,4,37,38 Determination of the thermal amplitude of the CA can be time consuming and is not necessary in all patients in clinical practice, but it can be useful for ruling out clinically insignificant CAs when relevant.39,40 We also routinely determine serum complement C3 and C4, although normal levels do not exclude CAD.1,40 Secondary CAS must be ruled out by clinical evaluation and, if required, appropriate imaging and relevant tests for specific infections.3,4 In my experience, computed tomography scans are unlikely to provide any additional information if there is no clinical suspicion of aggressive lymphoma or solid malignancy.
These examinations will establish the CAD diagnosis. In addition, the presence of CA-associated LPD should be evaluated by serum protein electrophoresis (SPEP), immune fixation, immunoglobulin class quantification, and bone marrow biopsy.4,23,32,33 If histologic evidence of clonal LPD is found, a mutational analysis of MYD status should be included. Flow cytometry in a bone marrow aspirate should also be performed; this examination is not sensitive enough if performed in peripheral blood.4,37 In studies, confirmation of clonal LPD has been achieved in up to 90% of cases, but this percentage will be lower in clinical practice, and negative findings do not exclude CAD.1,10,11 Furthermore, recognition of the specific features of CA-associated LPD is not always easy, and the biopsy findings may be interpreted as LPL, marginal zone lymphoma, or another indolent lymphoma, even in some cases of primary CAD.1,4,6 Centralized biopsy assessment by a pathology department with relevant expertise has been shown to markedly facilitate the recognition of CA-associated LPD.1
For CA titration and all immunoglobulin analyses, including SPEP and quantifications, it is critical to handle the blood samples as described in Table 1.33,38,41 Failure to comply with this procedure will result in reduced sensitivity and wrong estimates.
CAD: handling of samples
| Analysis | Material | Sampling | Handling of sample |
|---|---|---|---|
| Hb, blood cell counts | Blood | EDTA vacuum tube | Prewarm at 37-38°C before analysis if problems with agglutination |
| CA titer, thermal amplitude, immunoglobulin quantification, SPEP, immune fixation | Serum or plasma | Blood is drawn into prewarmed vacuum tubes (for serum, no gel or additive); place in warming cabinet or water bath at 37-38°C | Keep at 37-38°C until serum/plasma has been removed from clot/cells, after which sample can be handled at room temperature |
| Flow cytometry | Bone marrow aspirate (too low sensitivity if performed in peripheral blood) | Add EDTA or heparin | Prewarming before analysis will often be sufficient; if not, wash cells at 37-38°C; for description, see Ulvestad et al40 |
| Analysis | Material | Sampling | Handling of sample |
|---|---|---|---|
| Hb, blood cell counts | Blood | EDTA vacuum tube | Prewarm at 37-38°C before analysis if problems with agglutination |
| CA titer, thermal amplitude, immunoglobulin quantification, SPEP, immune fixation | Serum or plasma | Blood is drawn into prewarmed vacuum tubes (for serum, no gel or additive); place in warming cabinet or water bath at 37-38°C | Keep at 37-38°C until serum/plasma has been removed from clot/cells, after which sample can be handled at room temperature |
| Flow cytometry | Bone marrow aspirate (too low sensitivity if performed in peripheral blood) | Add EDTA or heparin | Prewarming before analysis will often be sufficient; if not, wash cells at 37-38°C; for description, see Ulvestad et al40 |
Reprinted with permission from Berentsen et al.41
To treat or not to treat, and to avoid ineffective therapies
Patients with mild anemia or compensated hemolysis and no clinical symptoms have not been shown to benefit from treatment. However, the restrictive attitude to drug therapy often found in the literature seems influenced by the poor efficacy of older therapies.9,42 Today, it is reasonable to treat those with bothersome symptoms who can be expected to benefit from therapy (ie, patients with symptomatic anemia or disabling Raynaud-like symptoms).4,33,43 Fatigue in the absence of significant anemia remains a more controversial indication, although there are emerging data on benefit.29 In unselected descriptive series, 70% to 80% of patients with CAD have received therapy.1,10,11 All patients, including those for whom pharmacologic treatment is not indicated, should be counseled on thermal protection, which has been described elsewhere.4,23,32,33 Counseling physicians and nurses on this rare disease seems equally important.
Not all treatments used in warm AIHA are effective in CAD. Unfortunately, ineffective therapies are still prevalent worldwide, as exemplified by case 1 and documented in several descriptive studies.1,10,11,44,45 Corticosteroids result in remission in <20% of patients, and in the few responders, unacceptably high maintenance doses are often required for sustained response.1,4,9,10,25,42 The incidence of adverse events (AEs), such as diabetes, skeletal events, and infection, is significant in patients with unspecified AIHA, although unknown in CAD.46 Therefore, CAD should not be treated with corticosteroids. Unspecific immunosuppressants are also associated with low response rates, although mainly older data are found in the literature.1,9,12,25,42 Because the extravascular hemolysis predominantly takes place in the liver,25,47 splenectomy is ineffective except in some rare cases of IgG-mediated disease or with a thermal amplitude approaching 37°C.1,42,48
Risk of thrombosis in CAD
Registry-based studies of patients diagnosed with CAD by their hospitals have found an increased risk of thromboembolic events (TEs).45,49,50 In the largest study, the relative risk was 3.1 as compared with matched controls.49 A large, hospital-based retrospective series of patients with verified CAD confirmed an increased risk, although apparently less marked.1 The largest unselected studies have not demonstrated any association with severity of anemia,1,49 but 1 study found a correlation with severity of hemolysis.49 Furthermore, a well-described cohort of selected, severely anemic patients did indicate a higher risk in this subgroup.26 In Europe and North America, the increased risk is most evident for venous TEs and less marked or absent for arterial TEs,1,49 whereas a recently presented Japanese study showed the opposite.45 Therefore, the fatal stroke in case 1 may be attributed to risk factors other than CAD. Successful CAD-directed therapy seems to reduce the frequency of TEs,26,29 but there are no data to show any impact of treatment of asymptomatic patients on TE risk.
As a reasonable preliminary conclusion, the author does not consider an assumed risk of TEs to be an indication for CAD-directed therapy in patients with mild anemia or compensated hemolysis. Untreated patients with severe anemia or acute exacerbation, as well as those with additional risk factors should probably receive prophylaxis with low-molecular-weight heparin or a direct oral anticoagulant.
Case 2
A woman in her early 80s, previously healthy apart from uncomplicated hypertension, was admitted because of a 4-month history of anemia. She had no ischemic symptoms but was mildly icteric. In the ward, she was perceived as being slightly cognitively reduced on some occasions, which was correctly attributed to her severe anemia. On admission, Hb was 6.2 g/dL, LDH was 647 U/L, bilirubin was 67 μmol/L, haptoglobin was <0.1 g/L, and reticulocytes were 118 × 109/L. IgM was 3.3 g/L, and monoclonal IgMκ was detected. DAT was strongly positive for C3d only, and her CA titer was 2048. Serum C3 was 0.54 g/L, and C4 was not detectable, consistent with massive complement consumption caused by classical pathway activation. In the bone marrow, flow cytometry revealed a ratio between κ+ and λ+ B cells of 26, and biopsy findings were consistent with CA-associated LPD. Clinically and radiologically, there were no enlarged lymph nodes or other signs of malignancy.
She was diagnosed with primary CAD. After informed consent, she was included in the Nordic bendamustine-rituximab trial43 and received 4 cycles of bendamustine at 90 mg/m2 on days 1 to 2 and rituximab at 375 mg/m2 on day 1 at 4-week intervals. Grade 2 to 3 neutropenia was noticed after each cycle, but she did not experience fever or infection. Hb started to rise after the second cycle, and she became transfusion independent. After completion of chemoimmunotherapy, she was clinically healthy and classified as having achieved a partial response (PR). Hb continued to improve and reached 13.8 g/L by 1 year posttherapy; markers of hemolysis were now completely normal, her monoclonal IgM had disappeared, no clonal LPD could be detected in her bone marrow, and she was reclassified as having achieved a complete response (CR). Six years posttherapy, she still seems healthy and has no anemia or hemolysis.
Therapy directed at the pathogenic B-cell clone
Although there are no randomized trials and no formal approval of any chemoimmunotherapy in CAD, remission after B cell–directed treatment has been confirmed in several systematic studies (Table 2). Two prospective, nonrandomized trials and some observational real-life studies have shown a beneficial effect of rituximab monotherapy.1,10,12,30 The prospective trials found a PR rate of ∼50%, few CRs, and a median response duration of 6.5 to 11 months (range, 2-42 months).12,30 Monotherapy with this monoclonal anti-CD20 antibody is generally well tolerated and has become the most commonly used documented therapy for CAD.32,51 Retreatment will often, but not always, succeed in relapsed patients.1,30 There are no published data on rituximab maintenance in CAD.
B cell–directed therapies effective in CAD
| Study/publication reference | Drug(s) studied | Study design | Patients/courses of therapy, n | OR, % | CR, % | Hb increase, g/dL* | Median response duration, mo | Toxicity† |
|---|---|---|---|---|---|---|---|---|
| 30 | Rituximab | Prospective, nonrandomized | 27/37 | 54 | 4 | 4.0 | 11 (observed) | Low |
| 12 | Rituximab | Prospective, nonrandomized | 20/20 | 45 | 5 | 3.1 | 6.5 (observed) | Low |
| 52 | Fludarabine + rituximab | Prospective, nonrandomized | 29/29 | 76 | 21 | 3.1 | >66 (estimated) | Significant |
| 43 | Bendamustine + rituximab | Prospective, nonrandomized | 45/45 | 71 | 40 | 4.0 | >32 (observed) | Relatively low, manageable |
| 53 | Bortezomib | Prospective, nonrandomized | 19/19 | 32 | 16 | 2.9 | 16 (observed) | Low |
| 1 | Bendamustine + rituximab | Follow-up, part of larger study | 45/45 | 78 | 53 | Not reevaluated | >88 (estimated) | Long-term: low |
| Study/publication reference | Drug(s) studied | Study design | Patients/courses of therapy, n | OR, % | CR, % | Hb increase, g/dL* | Median response duration, mo | Toxicity† |
|---|---|---|---|---|---|---|---|---|
| 30 | Rituximab | Prospective, nonrandomized | 27/37 | 54 | 4 | 4.0 | 11 (observed) | Low |
| 12 | Rituximab | Prospective, nonrandomized | 20/20 | 45 | 5 | 3.1 | 6.5 (observed) | Low |
| 52 | Fludarabine + rituximab | Prospective, nonrandomized | 29/29 | 76 | 21 | 3.1 | >66 (estimated) | Significant |
| 43 | Bendamustine + rituximab | Prospective, nonrandomized | 45/45 | 71 | 40 | 4.0 | >32 (observed) | Relatively low, manageable |
| 53 | Bortezomib | Prospective, nonrandomized | 19/19 | 32 | 16 | 2.9 | 16 (observed) | Low |
| 1 | Bendamustine + rituximab | Follow-up, part of larger study | 45/45 | 78 | 53 | Not reevaluated | >88 (estimated) | Long-term: low |
OR, overall response.
Median increase in responders.
Details provided in article text.
Addition of oral fludarabine, investigated in a prospective trial, resulted in a 76% response rate, with a 21% CR rate and a median increase in Hb by 3.1 g/dL (4.0 g/dL in those who achieved CR).52 The criteria for CR included disappearance of any histologic and flow cytometric signs of clonal bone marrow LPD. However, 41% of participants experienced grade 3 to 4 neutropenia, and infections were frequent. A later follow-up study seems to confirm a suspected risk of late-occurring malignancies but also shows a median response duration of 77 months and a 5-year continued response rate in 71% of responders, with >40% enjoying sustained response for 80 to 170 months.1,52
In a prospective trial published in 2017, 45 eligible patients received bendamustine at 90 mg/m2 on days 1 to 2 and rituximab at 375 mg/m2 on day 1 for 4 cycles at 4-week intervals.43 Seventy-one percent responded; 40% achieved CR and 31% PR. The median Hb increase was 4.4 g/dL in complete responders, 3.9 g/dL in those achieving PR, and 3.7 g/dL in the whole cohort. One-third of patients developed temporary grade 3 to 4 neutropenia, but only 11% had infections with or without neutropenia. Some patients experienced a long time to response (median, 1.9 months; range, 0.25-12 months) and even longer time to best response (median, 7 months; range, 1.5-30 months). Ninety percent of responders had not relapsed after 32 months.43 Follow-up data were published in 2020 as part of a larger descriptive study.1 Interestingly, these date showed a higher response rate at 78%, and 53% of participants had achieved CR. The median response duration could be estimated at >88 months, and 77% were still in remission after 5 years (Figure 3). The follow-up study also indicated that the bendamustine plus rituximab regimen is safe with regard to late-occurring malignancies.1

Kaplan-Meier plot showing probability of sustained remission in patients who responding to 4 cycles of rituximab plus bendamustine. Median response duration is not reached after 88 months, and estimated 5-year sustained response rate is 77%. Reprinted from Berentsen et al.1
Kaplan-Meier plot showing probability of sustained remission in patients who responding to 4 cycles of rituximab plus bendamustine. Median response duration is not reached after 88 months, and estimated 5-year sustained response rate is 77%. Reprinted from Berentsen et al.1
The occurrence of some very late responses and deepening of responses with time is probably best explained by the existence of long-lived, nonproliferating plasma cells that are more or less resistant to chemoimmunotherapy.6,54-56 These observations suggest that treatment should be discontinued after 4 cycles irrespective of remission status. Conversely, lack of response after 1 to 3 cycles should not lead to discontinuation of therapy. Furthermore, the response duration curve (Figure 3) seems to reach a plateau after ∼50 months, suggesting that a subset of patients will achieve very long-lasting remission, and some may even be cured.1
Bortezomib monotherapy was studied in a small, nonrandomized prospective trial in which 6 of 19 patients responded to 1 cycle of therapy (3 of 19 achieved CR; 3 of 19 achieved PR).53 CR criteria did not include regression of bone marrow findings, which was, however, achieved in most responders. Four of 6 responders maintained their remission for at least 16 months. These response rates may seem low but might be improved by using bortezomib in combinations and/or for an extended duration.
Case 3
An otherwise healthy woman in her 60s was referred to the outpatient hematology department because of anemia of 5-year duration. On examination, she had no acrocyanosis but admitted to moderate fatigue and mild cold-induced acral skin symptoms that prevented her from skiing in winter. She presented with an Hb level of 8.9 g/dL. Markers of hemolysis, DAT pattern, and SPEP were consistent with IgMκ-induced, complement-mediated hemolysis; her CA titer was 1028, she had no clinical evidence of malignancy, and she was diagnosed with CAD. Serum C4 was low (0.06 g/L). Bone marrow biopsy and flow cytometry were consistent with low-grade clonal B-cell expansion that was difficult to further classify. She was counseled on thermal protection but otherwise observed untreated.
During subsequent 6-year follow-up, there were no subjective changes, and Hb varied between ∼9.0 and ∼9.5 g/dL. Then, after a febrile urinary tract infection, Hb dropped to 7.8 g/dL; her anemia was symptomatic, and she received a transfusion. After resolution of the infection, Hb failed to rise markedly, and CAD-directed treatment was indicated, but she was reluctant to receive cytotoxic therapy. After informed consent, she was included in the Cardinal study, a nonrandomized phase 3 trial of sutimlimab in transfused patients with CAD.29 She received vaccination against meningococci, pneumococci, and Haemophilus influenzae B and was started on sutimlimab at 6.5 g IV every 14 days. Hb rose to 12.7 g/dL within 2 weeks on therapy, and her markers of hemolysis normalized, including a drop in bilirubin from 45 to 22 μmol/L by week 2 and to 14 μmol/L by week 4. Fourteen months after the initial response, she still receives sutimlimab every 14 days and tolerates the treatment well. Her Hb level is relatively stable at 12.9 to 14.1 g/dL; her fatigue has resolved, but her slight cold-induced circulatory symptoms persist.
Complement inhibitors in CAD
The first documentation of successful complement inhibition in IgM-mediated, complement-dependent AIHA was a case report describing therapy for CAD with the anti-C5 monoclonal antibody eculizumab,57 later followed by a prospective trial in 12 patients with CAD and 1 with severe CAS.26 However, C5 inhibition will not block the C3b-mediated, extravascular hemolysis, and therefore, not unexpectedly, efficient blockade of C5 resulted in only a small increase in Hb.26
Classical pathway inhibition should be expected to work better. A remarkable effect of high, frequent doses of plasma-derived C1 inhibitor has been demonstrated in a case of severe, IgM-mediated warm AIHA and later in a patient with CAS.58,59 Because patients with AIHA/CAD usually have sufficient endogenous C1 inhibitor production, C1 inhibitor therapy will require frequent and high doses and is probably not an attractive long-term approach.24
In a study published in 2014, the mouse monoclonal anti-C1s antibody TNT003 was shown to efficiently block classical complement pathway activation and opsonization of erythrocytes with C3b in an in vitro system with patient sera as a source of CA and normal human serum as a source of complement.22 These experiments led to the development of the corresponding humanized monoclonal antibody, sutimlimab (TNT009 or BIVV009).
In a clinical phase 1B trial, 7 of 10 patients with CAD responded to weekly doses of sutimlimab at 60 mg/kg with a rapid Hb increase of >2 g/dL (median, 3.9 g/dL).60 Elevated bilirubin levels normalized within 1 week in most participants. The results of a nonrandomized phase 3 study of sutimlimab in transfused patients were recently presented, showing similar findings in 24 patients treated with a fixed, biweekly dose of 6.5 or 7.5 g in those weighing <75 or ≥75 kg, respectively.29 This trial also found normalization of bilirubin and C4 levels by week 3 and marked improvement in fatigue within 1 week, as assessed by the Functional Assessment of Chronic Illness Therapy Fatigue scale. Participants were vaccinated against Neisseria meningitidis, Streptococcus pneumoniae, and H influenzae but did not receive prophylactic antibiotics. There were no meningococcal infections or other serious AEs related to the study drug.29
A randomized phase 3 trial of sutimlimab in nontransfused patients with CAD is ongoing but has finished inclusion (registered at www.clinicaltrials.gov as #NCT03347422). Furthermore, a safety and tolerability study of the anti-C1s antibody BIVV020 is now being conducted (registered at www.clinicaltrials.gov as #NCT04269551).
Complement modulation at the C3 level is also a promising approach. The C3 inhibitor pegcetacoplan (APL-2) is a pegylated cyclic peptide and a compstatin analog.61 Subcutaneous administration of pegcetacoplan was investigated in 2 phase 1 trials comprising a total of 51 healthy participants.62 Participants received vaccination as in the C1 inhibition trials. The pegcetacoplan studies found high efficacy in inhibiting the classical and alternative complement pathways, and the pharmacodynamic properties were favorable. There were few AEs and no serious AEs, although the maximal duration of administration was only 28 days. Phase 2 trials have found efficacy of pegcetacoplan in CAD as well as paroxysmal nocturnal hemoglobinuria.63
Future prospects
The B-cell–targeting approach has proved highly beneficial and might be further developed, aiming at equally efficacious and less toxic treatment options. Novel B-cell–targeting agents, such as Bruton tyrosine kinase (BTK) inhibitors, phosphatidylinositol 3-kinase δ inhibitors, and B-cell lymphoma 2 inhibitors, are highly active in other clonal B-cell LPDs and have a strong mechanistic rationale in CAD.64,65 No trial has been published, but remission after therapy with the BTK inhibitor ibrutinib has been observed in single cases.1 Systematic studies on the safety and efficacy of these drugs in CAD are warranted. In a recently presented retrospective series, 10 of 10 patients responded to the BTK inhibitor ibrutinib, with a median rise in Hb of 4.4 g/dL.66 All 7 transfused patients became transfusion independent. A phase 2 trial of the phosphatidylinositol 3-kinase δ inhibitor parsaclisib (INCB050465) is ongoing in patients with AIHAs of any type (registered at www.clinicaltrials.gov as #NCT03538041).
As mentioned, emergence of long-lived plasma cells may play a major role for the long time to response in some patients receiving B cell–directed chemoimmunotherapy and the modest but significant failure rate of these regimens.1,43,56 It is of interest, therefore, to explore the potential of efficient plasma cell–directed approaches as a supplement to these therapies in selected patients. Proteasome inhibitor–based combinations represent such a possibility.23,53 Furthermore, daratumumab, a monoclonal antibody against CD38 and powerful plasma cell–targeting agent, showed favorable effects in 2 patients with warm AIHA after stem cell transplantation.67,68 A recent case report described a rapid and convincing effect in a CAD patient with disabling agglutination-mediated ischemic symptoms and refractoriness to B cell–directed therapies.69 A small, systematic case series would be highly interesting.
Although the complement-directed therapies are still investigational, quite a lot of documentation has accumulated for sutimlimab.29,60,70 This drug will probably be available outside clinical trials in the near future. Additional clinical studies of pegcetacoplan are also warranted. Less documentation is available on other upstream complement inhibitors, such as the anti-C1q monoclonal antibody ANX005.71
Complement inhibition in CAD will have some limitations. The ischemic symptoms are not complement mediated and will not be relieved,72 as exemplified by case 3. Second, these therapies will have to be continued indefinitely to maintain their effect, as opposed to, for example, the 4-cycle bendamustine-rituximab regimen, which is temporary and highly efficacious.1,43 On the other hand, the upstream complement inhibitors are rapidly acting and probably relatively nontoxic.29,60,63 One would expect such therapies to be particularly helpful in severely anemic patients, those with acute exacerbations that do not resolve spontaneously, and those in whom chemoimmunotherapy has failed or is contraindicated. In some cases, complement inhibition may be used as a bridge to B cell–directed treatment.72
Conclusion
Appropriate diagnostic workup is essential for optimal therapy and must include monospecific DAT, CA titer, and relevant examinations for underlying bone marrow LPD. Treatment is not always indicated. Corticosteroids should not be used to treat CAD. Because there are no comparative trials, recommendations must be based on nonrandomized studies and will be influenced by personal experience. B cell–directed therapies are temporary and often highly efficacious; in the author’s opinion, they should remain the first choice in most cases. I prefer bendamustine plus rituximab over rituximab monotherapy in the first line for severely affected patients who are able to tolerate chemoimmunotherapy. In frail patients, rituximab monotherapy should be preferred. Sutimlimab is an emerging second-line option and may even be considered in the first line for initial treatment of severely anemic patients without disabling ischemic symptoms. Bortezomib-based regimens or, in highly selected cases, rituximab plus fludarabine will be alternatives in the second or third line. Several new options may appear in the near future.
Acknowledgments
The author thanks Tor Henrik Anderson Tvedt for providing some of the data on case 3. The author also thanks all colleagues who have contributed to the original studies on which much of this review is based and, not least, all patients who participated in these studies.
Authorship
Contribution: S.B. collected the information and wrote the article.
Conflict-of-interest disclosure: S.B. has received research support from Mundipharma; lecture honoraria from Apellis, Bioverativ (a Sanofi-Genzyme company), Janssen-Cilag, and True North Therapeutics; and consultancy and advisory board honoraria from Apellis, Bioverativ/Sanofi-Genzyme, and Momenta Pharmaceuticals.
Correspondence: Sigbjørn Berentsen, Haugesund Hospital, PO Box 2170, Haugesund, N-5504, Norway; e-mail: sigbjorn.berentsen@haugnett.no.
