

In this issue of Blood, 1 identify a novel role for fibroblast growth factor 23 (FGF-23), an osteocyte-derived hormone that regulates phosphate metabolism, in granulocyte colony-stimulating factor (G-CSF)–stimulated mobilization of hematopoietic stem and progenitor cells (HSPCs).
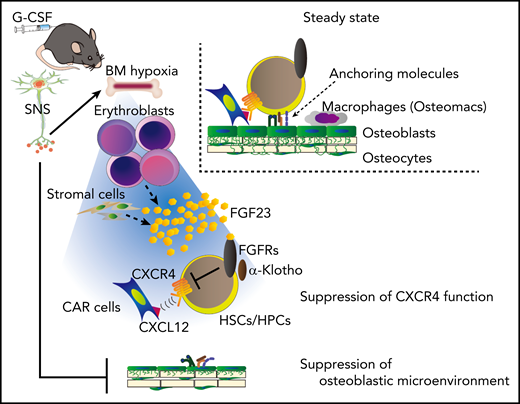
A model for G-CSF–induced mobilization by FGF-23 from erythroblasts. G-CSF or activation of the sympathetic nervous system, inducing hypoxia, increases the expression and release of FGF-23, mainly from erythroblasts (and also partially from stromal cells). Homeostatic mechanisms that contribute to HSPC anchoring to the microenvironment, such as chemoattraction toward CXCL12-abundant reticular (CAR) cells, are suppressed by the high concentration of bone marrow (BM) FGF-23, which counteracts the CXCR4 function via FGFRs. HSC, hematopoietic stem cell; SNS, sympathetic nervous system. See Figure 7 in the article by Ishii et al that begins on page 1457.
A model for G-CSF–induced mobilization by FGF-23 from erythroblasts. G-CSF or activation of the sympathetic nervous system, inducing hypoxia, increases the expression and release of FGF-23, mainly from erythroblasts (and also partially from stromal cells). Homeostatic mechanisms that contribute to HSPC anchoring to the microenvironment, such as chemoattraction toward CXCL12-abundant reticular (CAR) cells, are suppressed by the high concentration of bone marrow (BM) FGF-23, which counteracts the CXCR4 function via FGFRs. HSC, hematopoietic stem cell; SNS, sympathetic nervous system. See Figure 7 in the article by Ishii et al that begins on page 1457.
FGF-23 is a hormone secreted by osteoblasts and osteocytes that acts on the kidneys, parathyroid glands, heart, and bone. Now, as reported by Ishii et al, FGF-23 has been shown to play a role in G-CSF–mediated HSPC mobilization. Linkage analysis studies first identified the role of FGF-23 in phosphate wasting disorders.2 Targeted ablation of FGF-23 demonstrated its role in phosphate and vitamin D metabolism through its actions in the kidney and parathyroid glands.3 Intact FGF-23 binds with high affinity to a heterodimer of the membrane-bound FGF receptor 1 (FGFR-1) and Klotho proteins, activating canonical FGF-23 signaling.4
Noncanonical FGF signaling can also be mediated by other FGFRs independent of Klotho, particularly with extremely high FGF-23 levels, as in chronic kidney disease.4 A potential role of FGF-23 in hematopoiesis was initially suggested by increased red blood cell counts and elevated erythropoietin levels in mice lacking FGF-23. Consistent with an inhibitory role of FGF-23 in erythropoiesis, injection of FGF-23 in wild-type mice decreased erythropoiesis.5 Patients with chronic kidney failure have elevated levels of FGF-23, which may contribute to anemia by suppressing production of erythropoietin by the kidneys. However, direct effects of FGF-23 on erythropoiesis have been postulated, since the receptors activated by FGF-23, including FGFR-1, FGFR-3, and FGFR-4 and Klotho, are highly expressed in erythroid cells.4,5 Together these findings suggested that FGF-23 may contribute to suppression of erythropoiesis. On the other hand, data have shown that erythropoietin stimulates murine and human FGF-23 production not only in cells of the osteoblastic lineage but also in hematopoietic cells,6 specifically in erythropoietic precursor cells.5,7 Therefore, erythropoietin and FGF-23 may be components of a homeostatic mechanism linking erythropoiesis with skeletal and mineral metabolism.
The authors of the current study found increased levels of FGF-23 in the bone marrow extracellular fluid within the first 24 hours of G-CSF treatment. Since G-CSF induces sympathetic nervous system signals that impact mobilization, the pan β-adrenergic receptor agonist isoproterenol was tested. Isoproterenol also induced FGF-23. Surprisingly, administration of G-CSF or isoproterenol increased FGF-23 expression in erythroblastic cells, identified by flow cytometry as CD45−Ter119+CD51+ cells. Hypoxia, which is known to be induced by G-CSF in the bone marrow microenvironment, also increased FGF-23 in an erythroblastic cell line. Next, mice with a global deletion of FGF-23, previously found to have skeletal fragility and abnormal mineral metabolism,3 were shown to have a decrease in G-CSF–dependent mobilization of HSPCs. Notably, chimeric mice where FGF-23−/− bone marrow was transplanted in wild-type recipients also had inhibition of G-CSF–dependent stem cell mobilization, suggesting that hematopoietic cells (likely erythroblastic cells) are a critical source of FGF-23 responding to G-CSF mobilization. Mice with targeted deletion of FGF-23 in osteocytes had no reduction in G-CSF–induced stem cell mobilization, although a contribution of osteoblastic FGF-23 could not be excluded using this model.
Next, to investigate the mechanism of this effect of FGF-23 on HSPC mobilization, the authors evaluated CXCL12, the critical cytokine regulating the retention of HSCs in the bone marrow. Mice lacking FGF-23 globally, or in the bone marrow, demonstrated no change in the levels of CXCL12. This suggests that the effect of FGF-23 in stem cell mobilization is not due to modulation of CXCL12 expression. However, FGF-23 did inhibit HSPC migration toward CXCL12, and pharmacologic studies suggested that this may be due to FGF-23–dependent signaling and not the modulation of CXCL12 binding to its receptor.
This novel role of erythroblastic FGF-23 in modulating G-CSF–mediated mobilization may have clinical implications in diseases or iatrogenic conditions in which erythroblasts are decreased. For example, the lack of local FGF-23 due to low erythroblastic numbers in patients with multiple myeloma or patients treated with chemotherapy could contribute to insufficient mobilization after G-CSF. It will also be important to test whether high circulating levels of FGF-23 in chronic kidney disease may impair HSPC retention in the bone marrow. Notably, ongoing phase 2 and 3 studies are evaluating safety and efficacy of burosomab, a fully human monoclonal antibody that inhibits FGF-23, in patients with FGF-23–induced hypophosphatemic rickets/osteomalacia.8,9 The current work suggests that the hematopoietic consequences of these treatments need to be investigated.
A number of specific questions about the role of FGF-23 in hematopoiesis remain to be addressed. First, additional studies, with targeting of FGF-23 deletion to erythroid populations may further clarify the physiologic role of FGF-23 in erythroblastogenesis and HSPC retention in bone marrow. Moreover, the mechanisms by which FGF-23 regulates mobilization of HSPCs are not clearly elucidated here and should be further explored. Finally, it is unclear if or how this novel action of FGF-23 in regulation of HSPCs contributes to its role in anemia associated with inflammation and chronic kidney disease.
In summary, Ishii and colleagues have delineated the novel role of FGF-23, a mediator of mineral metabolism, in mobilization of hematopoietic stem cells. They have provided persuasive data supporting the importance of local production of FGF-23 by erythroblasts in response to sympathetic signals and G-CSF (see figure). This work adds erythroblasts to the list of hematopoietic cells that may have a regulatory role in the hematopoietic stem cell niche. These novel data also provide additional support to the concept that skeletal and hematopoietic remodeling are linked, demonstrating once more the reciprocal and coordinated regulation of the skeleton and the hematopoietic marrow.
Conflict-of-interest disclosure: The author declares no competing financial interests.
