

Key Points
SMOC1 interacts with thrombin to increase its activity and regulate platelet function.
Platelets from diabetic subjects demonstrate elevated SMOC1 expression and hyperreactivity, which are abrogated upon SMOC1 neutralization.

Abstract
Secreted modular calcium-binding protein 1 (SMOC1) is an osteonectin/SPARC-related matricellular protein, whose expression is regulated by microRNA-223 (miR-223). Given that platelets are rich in miR-223, this study investigated the expression of SMOC1 and its contribution to platelet function. Human and murine platelets expressed SMOC1, whereas platelets from SMOC1+/− mice did not present detectable mature SMOC1 protein. Platelets from SMOC1+/− mice demonstrated attenuated responsiveness to thrombin (platelet neutrophil aggregate formation, aggregation, clot formation, Ca2+ increase, and β3 integrin phosphorylation), whereas responses to other platelet agonists were unaffected. SMOC1 has been implicated in transforming growth factor-β signaling, but no link to this pathway was detected in platelets. Rather, the SMOC1 Kazal domain directly bound thrombin to potentiate its activity in vitro, as well as its actions on isolated platelets. The latter effects were prevented by monoclonal antibodies against SMOC1. Platelets from miR-223–deficient mice expressed high levels of SMOC1 and exhibited hyperreactivity to thrombin that was also reversed by preincubation with monoclonal antibodies against SMOC1. Similarly, SMOC1 levels were markedly upregulated in platelets from individuals with type 2 diabetes, and the SMOC1 antibody abrogated platelet hyperresponsiveness to thrombin. Taken together, we have identified SMOC1 as a novel thrombin-activating protein that makes a significant contribution to the pathophysiological changes in platelet function associated with type 2 diabetes. Thus, strategies that target SMOC1 or its interaction with thrombin may be attractive therapeutic approaches to normalize platelet function in diabetes.
Introduction
Platelets are anuclear cell fragments derived from megakaryocytes that play an essential role in primary hemostasis and clot formation. Although the latter function is essential, only a small percentage of circulating platelets are ever included in a thrombus. It is now generally appreciated that platelets play a much broader role in cardiovascular homeostasis. For example, platelets are a source of numerous bioactive factors, including sphingosine-1–phosphate,1 adenosine triphosphate,2 and transforming growth factor-β (TGF-β)3 ; chemokines, such as CCL54 ; and a spectrum of RNA species, including circular RNAs5 and microRNAs.6,7 One platelet-enriched microRNA is microRNA-223 (miR-223),6-8 the levels of which are decreased in platelets from diabetic individuals as a consequence of the activation of calpain and cleavage of Dicer.9 This led to the altered expression of miR-223 targets (eg, factor XIIIA and β1 integrin) and resulted in platelet hyperreactivity.9 A decrease in platelet miR-223 levels has also been linked to the enhanced incidence of cardiovascular complications associated with diabetes.10-12
Secreted modular calcium-binding protein 1 (SMOC1) is a matricellular protein that is localized to the basement membrane of different tissues13-16 that was recently identified as an miR-223 target.17 Although the physiological functions of SMOC1 are not fully understood, the protein has been associated with osteoblast differentiation, ocular and limb development, and angiogenesis.16-18 There have been clear links between mutations in SMOC1 and Waardenburg anophthalmia syndrome,19,20 and some studies have shown possible involvement in cancer.21,22 Functionally, SMOC1 has been associated with antagonism of bone morphogenetic protein and TGF-β signaling (for a review, see Bradshaw15 ). In the vasculature, SMOC1 was reported to bind to endoglin to inhibit TGF-β–induced activin receptor-like kinase 5 (ALK5) signaling and promote angiogenesis in vitro and in vivo17 ; otherwise, little is known about the cardiovascular role of SMOC1. Therefore, the aim of this investigation was to determine whether SMOC1 is expressed in platelets and, if so, to determine its role in the regulation of platelet function, particularly in diabetes.
Methods
Reagents
Thrombin was from Haemochrom Diagnostica (Essen, Germany), type I collagen was from BD Transduction Laboratories (Heidelberg, Germany), and U46619 was from Enzo Life Science (Lörrach, Germany). The ALK5 inhibitor 2-(5-chloro-2-fluorophenyl)-N-(pyridin-4-yl)pteridin-4-amine (SD-208) was from Calbiochem (Darmstadt, Germany). The protease-activated receptor 3 (PAR3; SFNGGP-NH2) and PAR4 (AYPGKF-NH2) agonists were from AnaSpec (Fremont, CA). The anti-SMOC1 and anti–integrin-β1 antibodies were from Abnova (Aachen, Germany), anti-Tyr773 β3 integrin was from Thermo Fisher Scientific (Waltham, MA), and the anti-β3 integrin antibody was from Epitomics (London, United Kingdom). The PAR-1, PAR-3, and CD144 antibodies were from Santa Cruz Biotechnology (Dallas, TX), the PAR-4 antibody was from Alomone Labs (Jerusalem, Israel), and the β-actin antibody was from Sigma-Aldrich (Munich, Germany). Recombinant human and mouse SMOC1 were from R&D Systems (Minneapolis, MN).
Animals
Wild-type (WT; C57BL/6) mice were from Charles River (Sulzfeld, Germany), SMOC1 [B6D2-Smoc1 < Tn(sb-lacZ,GFP)PV384Jtak>/JtakRbrc] mice (SMOC1+/−) were from the RIKEN BioResource Center (Tsukuba, Japan), and miR-223–knockout mice (miR223y/−)23 were provided by Fernando Camargo (Boston, MA). All animals were housed in conditions that conform to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (publication no. 85-23). The University Animal Care Committee and the Federal Authority for Animal Research at Regierungspräsidium Darmstadt (Hessen, Germany) approved the study protocol (#F28/21 and #F28/36). Age-, sex-, and strain-matched animals (littermates) were used throughout the studies. For the isolation of blood, mice were euthanized using 4% isoflurane in air and subsequently exsanguinated.
Diabetic study population
A total of 16 patients (mean age 43.5 years; range, 30-60) with type 2 diabetes mellitus attending the Endokrinologikum Frankfurt Clinic for routine control visits were included in the study with the following parameters: hemoglobin A1c >7.4% (9.01 ± 0.37%) and fasting plasma glucose, 8.35 ± 0.82 mmol/L. Nine patients were treated with insulin in combination with metformin, 1 was treated with metformin and liraglutide, 4 were with treated with insulin alone, and 2 were not treated. Nondiabetic age-matched subjects (mean age, 41.2 years; range, 25-60; hemoglobin A1c, 4.98 ± 0.58%; fasting plasma glucose, 5 ± 0.19 mmol/L) who had not taken any medication known to interfere with platelet aggregation for ≥10 days before the experiment served as the control group. The study protocol was approved by the Goethe University Hospital Ethics Committee (No. E 61/09 Geschäfts Nr 86/09), and all of the participants gave written informed consent.
Platelet isolation, adhesion, spreading, aggregation, and clot retraction
Intracellular Ca2+ measurement
Fibrin clot formation
The formation of fibrin clots in vitro was assessed as described,27 using fibrinogen (0.27 mg/mL) and thrombin (0.1 U/mL). Clot formation was assessed by a change in turbidity (ie, absorbance at λ350 nm using the EnVision Multilabel Plate Reader; PerkinElmer, Waltham, MA).
Immunoblotting
Endothelial cells and platelets were lysed in Triton X-100 buffer enriched with protease and phosphatase inhibitors. Samples were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and subjected to western blotting; detection was performed by enhanced chemiluminescence using a commercially available kit (Amersham, Freiburg, Germany) as described.28
Thrombin activity assay
Thrombin activity was measured using a commercially available kit and performed according to the manufacturer’s instructions (MAK242; Sigma-Aldrich).
Microscale thermophoresis assay
Thrombin was labeled with amilamide fluorescent dye using a Monolith Protein Labeling Kit (NanoTemper Technologies, Munich, Germany). Assays were carried out in 20 mmol/L Tris buffer, pH 7.5. For the direct binding assay, 10 µL of labeled protein was mixed with 10 µL of recombinant SMOC1 and incubated at room temperature for 1 hour. The sample was loaded in a capillary and then heated with an infrared laser. The change in sample fluorescence upon laser activation was monitored by calculating the ratio between the fluorescence before the T-jump (infrared laser off, t = 30 seconds) and prior to the infrared laser activation (t = 0 seconds). Data were analyzed using NanoTemper analysis software. Dissociation constants (Kd) and Hill coefficients values were determined using “T-jump + Thermophoresis” settings and GraphPad Prism 7.
Mutagenesis studies
Plasmids containing the full-length SMOC1 (SMOC1 FL), SMOC1ΔKazal (SMOC1Δ108-268 bp), and the isolated Kazal domain (108-268 bp) were synthetized de novo and cloned into the PS100020-pCMV6-AC between the restriction sites 5′ SgfI and 3′ MluI by Blue Heron Biotech (Bothell, WA). A431 cells were transfected with Lipofectamine 3000 (Invitrogen, Karlsruhe, Germany), according to the manufacturer’s instructions, using 5 µg of plasmid per million cells. The medium was changed after 6 hours, and cells were cultured for an additional 48 hours in the presence of serum, followed by 24 hours in serum-free medium. Thereafter, the culture medium was collected and concentrated using Amicon Ultra Filters (15 mL; Merck, Darmstadt, Germany). Concentrations of full-length SMOC1 and the SMOC mutants were determined using a specific enzyme-linked immunosorbent assay (LS-F52641; LSBio, Seattle, WA).
Endothelial cell culture
Human umbilical vein endothelial cells were isolated and cultured as described previously29 and used up to passage 2. The use of human material in this study complies with the principles outlined in the Declaration of Helsinki,30 and the isolation of endothelial cells was approved in written form by the Goethe University Ethics Committee. To silence SMOC1 expression, endothelial cells were transfected with small interfering RNA (siRNA) directed against SMOC1 (sense: 5′-UUG UUA AUG UCG UUG CUG CdTdT-3′ and antisense: 5′-GCA GCA ACG ACA UUA ACA UUA ACA AdTdT-3′) or with control oligonucleotides (both from Eurogentec, Cologne, Germany). All transfections were performed using the Lipofectamine RNAiMax Transfection Kit (Invitrogen, Darmstadt, Germany), according to the manufacturer’s instructions.
Endothelial cell permeability
Human endothelial cells were treated with siRNA directed against SMOC1 and seeded onto tissue culture inserts (1-µm pore size; Sarstedt AG & Co KG, Nümbrecht, Germany). When confluent, fluorescein isothiocyanate-dextran (average molecular mass 60 000-76 000 Da; 1 mg/mL) was added to the upper chamber, followed by thrombin (0.1 U/mL) treatment. Medium from the lower chamber was recovered after 5 and 15 minutes, and fluorescence was measured immediately (λ excitation 490 mm; λ emission 520 nm).
Immunofluorescence
Following treatment with thrombin (0.3 U/mL) in the absence and presence of recombinant SMOC1, endothelial cells were fixed with formalin, permeabilized with Triton X-100 (0.1%), blocked with 3% bovine serum albumin, and incubated with primary antibodies overnight at 4°C. Thereafter, anti-goat and anti-mouse secondary antibodies (1:200 in phosphate-buffered saline) were added. After washing, samples were incubated with DAPI (10 ng/mL), washed and mounted in Fluoromount-G (Thermo Fisher Scientific). Images were taken using a confocal microscope (LSM-780; Zeiss, Jena, Germany) using ZEN software (Zeiss).
Statistics
Data are expressed as mean ± standard error of the mean. Statistical evaluation was performed using the Student t test for unpaired and paired data and 1- and 2-way analysis of variance (ANOVA), followed by a Bonferroni posttest or ANOVA, for repeated measures, where appropriate. Values of P < .05 were considered statistically significant.
Results
SMOC1 expression and function in platelets
Platelets from WT mice expressed the premature intracellular form of SMOC1, as well as the mature secreted protein (Figure 1A). SMOC1−/− mice are available, but they usually die shortly after birth18,19 ; therefore, platelet reactivity was studied in WT (SMOC1+/+) and SMOC1+/− littermates. This was justified, because the expression of the mature SMOC1 protein was barely detectable in platelets from heterozygous mice (Figure 1A). There were no detectable differences in platelet counts or blood cell counts in samples from WT and SMOC+/− mice (supplemental Figure 1, available on the Blood Web site). Although there was no effect of SMOC1 deficiency on platelet adherence to collagen, SMOC1-deficient platelets failed to spread (Figure 1B). Moreover, SMOC1+/− mice showed decreased numbers of thrombin-induced platelet leukocyte aggregates in whole blood (Figure 1C). SMOC1-deficient platelets also demonstrated a significantly attenuated aggregation in response to thrombin (Figure 1D), as well as a defect in clot retraction (Figure 1E).
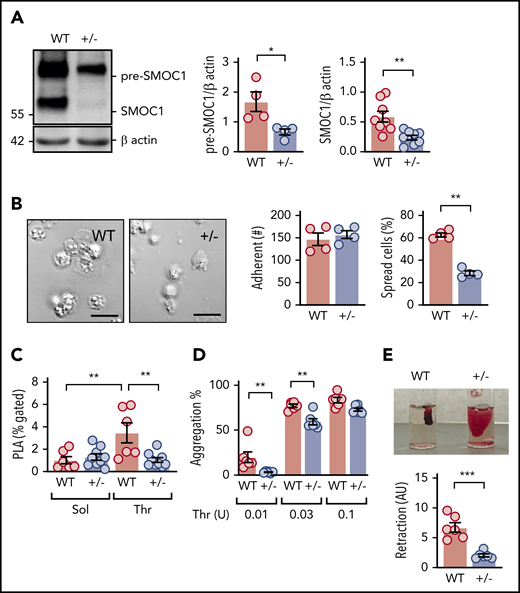
SMOC1 deficiency decreases platelet function in murine platelets. (A) Expression of the premature (pre-SMOC1) and mature secreted (SMOC1) forms of SMOC1 in platelets from WT and SMOC1+/− (+/−) littermates; n = 6 animals per group. *P < .05, **P < .01, Student t test. (B) Adherent and nonspread (inactive) platelets isolated from WT and SMOC1+/− (+/−) mice after seeding onto collagen; n = 4 animals per group. Scale bars, 10 μm. **P < .01, Student t test. (C) Platelet-leukocyte aggregates (PLA) in isolated whole blood from WT and SMOC1+/− (+/−) mice following treatment with solvent (Sol) or thrombin (Thr); n = 6 animals per group. **P < .01, 2-way ANOVA and Newman-Keuls test. (D) Thrombin (Thr)-induced platelet aggregation in washed platelets from WT and SMOC1+/− (+/−) mice; n = animals 7 per group. **P < .01, 2-way ANOVA and Bonferroni test. (E) Thrombin-induced clot retraction using erythrocyte-spiked platelet-rich plasma from WT and SMOC1+/− (+/−) mice; n = 6 animals per group. ***P < .001, Student t test.
SMOC1 deficiency decreases platelet function in murine platelets. (A) Expression of the premature (pre-SMOC1) and mature secreted (SMOC1) forms of SMOC1 in platelets from WT and SMOC1+/− (+/−) littermates; n = 6 animals per group. *P < .05, **P < .01, Student t test. (B) Adherent and nonspread (inactive) platelets isolated from WT and SMOC1+/− (+/−) mice after seeding onto collagen; n = 4 animals per group. Scale bars, 10 μm. **P < .01, Student t test. (C) Platelet-leukocyte aggregates (PLA) in isolated whole blood from WT and SMOC1+/− (+/−) mice following treatment with solvent (Sol) or thrombin (Thr); n = 6 animals per group. **P < .01, 2-way ANOVA and Newman-Keuls test. (D) Thrombin (Thr)-induced platelet aggregation in washed platelets from WT and SMOC1+/− (+/−) mice; n = animals 7 per group. **P < .01, 2-way ANOVA and Bonferroni test. (E) Thrombin-induced clot retraction using erythrocyte-spiked platelet-rich plasma from WT and SMOC1+/− (+/−) mice; n = 6 animals per group. ***P < .001, Student t test.
SMOC1 deficiency in thrombin-induced platelet signaling
The differences in platelet reactivity were also reflected in the downstream tyrosine phosphorylation of β3-integrin; although thrombin induced the phosphorylation of β3-integrin (on Tyr773) in platelets from WT mice, this response was largely absent in platelets from SMOC1-deficient animals (Figure 2A; supplemental Figure 2). However, the thrombin-induced phosphorylation of β3-integrin was restored by the addition of recombinant SMOC1 protein (Figure 2B). To determine whether the effects observed were specific to thrombin, platelet aggregation in response to other agonists was assessed. Intriguingly, the platelet aggregation and the tyrosine phosphorylation of β3-integrin elicited by collagen, the thromboxane analog U46619, and adenosine diphosphate were comparable in WT and SMOC1+/− mice (supplemental Figure 3). These findings implied that the lack of SMOC1 specifically affected responses to thrombin.
![Effects of SMOC1 on thrombin-dependent responses in murine platelets. (A) Thrombin-induced phosphorylation of β3-integrin (on Tyr773; pY773) in platelets from WT and SMOC1+/− (+/−) mice; n = 5 animals per group. **P < .01, ANOVA and Bonferroni test. (B) Effect of thrombin (Thr; 0.1 U/mL) on β3-integrin phosphorylation (on Tyr773; pY773) in platelets from SMOC1+/− (+/−) mice pretreated with solvent (Sol) or murine recombinant SMOC1 (rSMOC1; 10 µg/mL, 45 minutes); n = 8 per group. ***P < .001, 2-way ANOVA and Bonferroni test. (C) PAR3 and PAR4 receptor expression in platelets from WT and SMOC1+/− (+/−) mice; n = 4 animals per group. (D) Basal and agonist-induced increases in platelet Ca2+ levels ([Ca2+]i) in platelets from WT and SMOC1+/− (+/−) littermates stimulated with thrombin (Thr; 1 U/mL), a PAR3 agonist (100 µM), or a PAR4 agonist (100 µM); n = 4 animals per group. ***P < .001, 2-way ANOVA and Bonferroni test.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/12/10.1182_blood.2020009405/1/m_bloodbld2020009405f2.png?Expires=1763592109&Signature=hQeoUbVSNNFvx7StEJIh1zzddjXs1VfGFB2~ErJWIgSiC5B6H7AugbFXpb48Qe~mtwndfErE6EKoYlEuoOqOhk93XaIdgsDTw5RDUGDtN4vaMq6P2Rq5D2aD62J7H0f8gCCSFIL2k0xN-hAcvD6ML1bNjairc5DAR7jPV0B51fCOLVa96PiB8ob-FdRQkqDcoublTumOV3McIchBjA8nOfS6OiMVjJxDGBKHP-kJ8EHeztF7f~FuPZBgTgbHEy~iQcZ6RrJTkcuPhPDZjPLwFoHOGjNerljC~OKjC~P3-d0DZjm03UvzlWcRhV22EOZzbVOa2laDwibodb~1TP4yvA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Effects of SMOC1 on thrombin-dependent responses in murine platelets. (A) Thrombin-induced phosphorylation of β3-integrin (on Tyr773; pY773) in platelets from WT and SMOC1+/− (+/−) mice; n = 5 animals per group. **P < .01, ANOVA and Bonferroni test. (B) Effect of thrombin (Thr; 0.1 U/mL) on β3-integrin phosphorylation (on Tyr773; pY773) in platelets from SMOC1+/− (+/−) mice pretreated with solvent (Sol) or murine recombinant SMOC1 (rSMOC1; 10 µg/mL, 45 minutes); n = 8 per group. ***P < .001, 2-way ANOVA and Bonferroni test. (C) PAR3 and PAR4 receptor expression in platelets from WT and SMOC1+/− (+/−) mice; n = 4 animals per group. (D) Basal and agonist-induced increases in platelet Ca2+ levels ([Ca2+]i) in platelets from WT and SMOC1+/− (+/−) littermates stimulated with thrombin (Thr; 1 U/mL), a PAR3 agonist (100 µM), or a PAR4 agonist (100 µM); n = 4 animals per group. ***P < .001, 2-way ANOVA and Bonferroni test.
Effects of SMOC1 on thrombin-dependent responses in murine platelets. (A) Thrombin-induced phosphorylation of β3-integrin (on Tyr773; pY773) in platelets from WT and SMOC1+/− (+/−) mice; n = 5 animals per group. **P < .01, ANOVA and Bonferroni test. (B) Effect of thrombin (Thr; 0.1 U/mL) on β3-integrin phosphorylation (on Tyr773; pY773) in platelets from SMOC1+/− (+/−) mice pretreated with solvent (Sol) or murine recombinant SMOC1 (rSMOC1; 10 µg/mL, 45 minutes); n = 8 per group. ***P < .001, 2-way ANOVA and Bonferroni test. (C) PAR3 and PAR4 receptor expression in platelets from WT and SMOC1+/− (+/−) mice; n = 4 animals per group. (D) Basal and agonist-induced increases in platelet Ca2+ levels ([Ca2+]i) in platelets from WT and SMOC1+/− (+/−) littermates stimulated with thrombin (Thr; 1 U/mL), a PAR3 agonist (100 µM), or a PAR4 agonist (100 µM); n = 4 animals per group. ***P < .001, 2-way ANOVA and Bonferroni test.
Thrombin elicits its effects by activating the PARs, and murine platelets express PAR3 and PAR4,31 both of which were expressed at similar levels in platelets from WT and SMOC+/− littermates (Figure 2C). However, although the Ca2+ response to thrombin was clearly attenuated in platelets lacking SMOC1, responses to the specific PAR3 agonist SFNGGP-NH2 and the PAR4 agonist AYPGKF-NH2 were comparable in SMOC1-expressing and -deficient platelets (Figure 2D). Platelets contain large amounts of TGF-β,3 and cross talk between TGFβ/ALK5 and PAR2/PAR1 has been proposed in some cells.32,33 However, ALK5 inhibition had no effect on the thrombin-induced phosphorylation of β3-integrin (supplemental Figure 4). These results suggest that the impaired platelet function in SMOC1-deficient mice was specific to thrombin but could not be accounted for by altered thrombin receptor expression or activation.
Identification of SMOC1 as a thrombin-binding protein
SMOC1 has been reported to bind cell-associated proteins, including laminins,14 C-reactive protein, fibulin-1, vitronectin,34 heparin and heparan sulfate,35 tenascin-C,22 and proepidermal growth factor.36 Therefore, we hypothesized that SMOC1 was able to bind to thrombin. In vitro activity assays revealed that recombinant SMOC1 increased thrombin activity, an effect reversed by the addition of a monoclonal antibody directed against SMOC1 (Figure 3A). Similarly, in washed human platelets from healthy volunteers, the SMOC1 antibody attenuated the aggregation induced by low concentrations of thrombin (Figure 3B), as well as clot retraction (Figure 3C). A monoclonal antibody directed against β-actin was without effect. A direct interaction between recombinant SMOC1 and thrombin was demonstrated in a microscale thermophoresis assay by a shift in the fluorescence decay of labeled thrombin in the presence of increasing concentrations of SMOC1 (Figure 3D), which gave a Kd of 1.46 ± 0.5 µmol/L. No decay was observed following the denaturation of SMOC1.
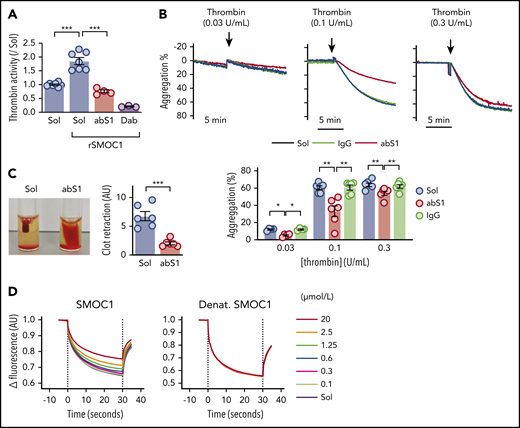
Interaction between SMOC1 and thrombin. (A) Thrombin activity in the presence of solvent (Sol) or recombinant human SMOC1 (rSMOC1) and in the absence and presence of antibodies directed against SMOC1 (abS1). The thrombin inhibitor dabigatran (Dab) was included as control; n = 3 to 7 independent experiments. ***P < .001, 1-way ANOVA and Bonferroni test. (B) Effect of a SMOC1 antibody (abS1; 0.625 ng/µL) on the thrombin-induced aggregation of washed human platelets. Immunoglobulin G (IgG) was included as control; n = 6 independent donors. *P < .05, **P < .01, 2-way ANOVA and Bonferroni test. (C) Thrombin-induced (0.1 U/mL) clot retraction in platelet-rich plasma (3 × 108 platelets per mL), in the presence of calcium (20 mmol/L) and 10 µL of erythrocytes to enhance the contrast of the clot. The clots were allowed to retract for up to 1 hour; n = 6 independent donors. ***P < .001, Student t test. (D) Microscale thermophoresis assay for the interaction between fluorescently labeled thrombin and recombinant SMOC1 or heat-denatured (Denat.) recombinant SMOC1 (0.1-20 µmol/L). The dotted lines indicate the beginning and the end of the laser pulse; n = 3 independent experiments. AU, arbitrary units.
Interaction between SMOC1 and thrombin. (A) Thrombin activity in the presence of solvent (Sol) or recombinant human SMOC1 (rSMOC1) and in the absence and presence of antibodies directed against SMOC1 (abS1). The thrombin inhibitor dabigatran (Dab) was included as control; n = 3 to 7 independent experiments. ***P < .001, 1-way ANOVA and Bonferroni test. (B) Effect of a SMOC1 antibody (abS1; 0.625 ng/µL) on the thrombin-induced aggregation of washed human platelets. Immunoglobulin G (IgG) was included as control; n = 6 independent donors. *P < .05, **P < .01, 2-way ANOVA and Bonferroni test. (C) Thrombin-induced (0.1 U/mL) clot retraction in platelet-rich plasma (3 × 108 platelets per mL), in the presence of calcium (20 mmol/L) and 10 µL of erythrocytes to enhance the contrast of the clot. The clots were allowed to retract for up to 1 hour; n = 6 independent donors. ***P < .001, Student t test. (D) Microscale thermophoresis assay for the interaction between fluorescently labeled thrombin and recombinant SMOC1 or heat-denatured (Denat.) recombinant SMOC1 (0.1-20 µmol/L). The dotted lines indicate the beginning and the end of the laser pulse; n = 3 independent experiments. AU, arbitrary units.
The SMOC1 protein contains several domains, including a follistatin-like domain, 2 thyroglobulin-like domains, 1 extracellular calcium-binding domain, and 1 domain unique to the SMOC proteins (Figure 4A). The follistatin-like domain is composed of 2 subdomains, with the second being similar to the Kazal domain that characterizes serine proteinase inhibitors but is different enough to be classed as a “nonclassical” Kazal domain.14 To determine the potential site of binding of thrombin to SMOC1, the supernatants were recovered from A431 cells expressing the full-length SMOC1 protein, a mutant lacking the Kazal domain (ΔKazal), or the isolated Kazal domain, and the concentration of the SMOC1 proteins was determined by specific enzyme-linked immunosorbent assay. The binding of full-length SMOC1 to thrombin was somewhat better than that of the commercially available recombinant SMOC1 (Kd = 2.15 ± 0.2 nmol/L), and the deletion of the Kazal domain markedly attenuated this interaction (Kd = 76.8 ± 0.3 nmol/L). However, the isolated Kazal domain was almost as effective at binding thrombin as the full-length protein (Kd = 7.7 ± 0.2 nmol/L) (Figure 4B). Consistent with these findings, the activity of thrombin was increased by full-length SMOC1 and the isolated Kazal domain but not by the ΔKazal mutant (Figure 4C). Similarly, the thrombin-induced aggregation of washed human platelets was increased by full-length SMOC1 and the Kazal domain but not by the ΔKazal SMOC1 mutant (Figure 4D). Moreover, in a purely in vitro situation, thrombin-induced fibrin clot formation was potentiated by full-length SMOC1 and the Kazal peptide but not by ΔKazal SMOC1 (Figure 4E).
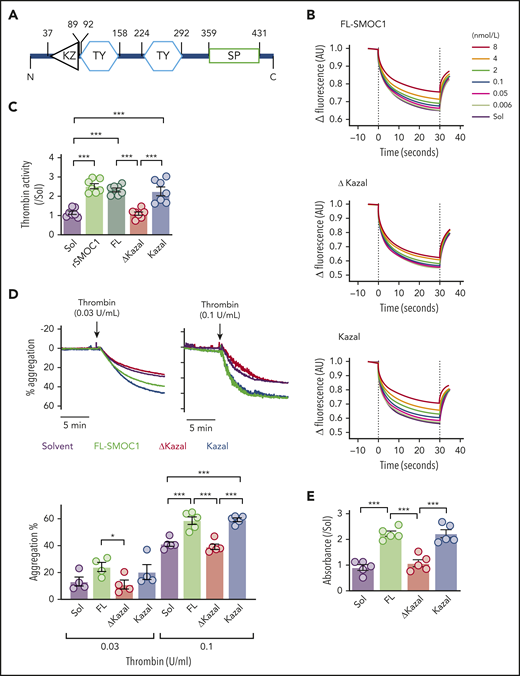
Identification of the thrombin-binding domain of SMOC1. (A) Schematic diagram of the SMOC1 protein and the SMOC1 antibody binding site. KZ, Kazal domain; SP, SPARC domain; TY, thyroglobulin domain. (B) Microscale thermophoresis assay for the interaction between fluorescently labeled thrombin and full-length (FL) SMOC1 (FL-SMOC1), SMOC1ΔKazal, (ΔKazal), or the SMOC1 Kazal (Kazal) domain (6 pmol/L-8 nmol/L). The dotted lines indicate the beginning and the end of the laser pulse; n = 4 or 5 independent experiments. (C) Thrombin activity assay in the presence of solvent (Sol; A431 cell supernatant), recombinant SMOC1 (rSMOC1), the overexpressed full-length SMOC1 protein (FL), SMOC1ΔKazal (ΔKazal), and the isolated Kazal domain (Kazal) (all 4 nmol/L); n = 6. ***P < .001, 1-way ANOVA and Bonferroni. (D) Thrombin-induced aggregation of washed human platelets in the presence of Sol (A431 cell supernatant), full-length SMOC1 (FL-SMOC1), the ΔKazal mutant, and the isolated Kazal domain (all 4 nmol/L); n = 5 donors. *P < .05, ***P < .001, 2-way ANOVA and Bonferroni test. (E) Fibrin clot formation (absorbance λ350 nm) in vitro induced by thrombin (0.1 U/mL) in the presence of Sol, full-length SMOC1 (FL), the ΔKazal mutant, and the isolated Kazal domain (all 4 nmol/L); n = 5 independent experiments. ***P < .001, 1-way ANOVA and Bonferroni test.
Identification of the thrombin-binding domain of SMOC1. (A) Schematic diagram of the SMOC1 protein and the SMOC1 antibody binding site. KZ, Kazal domain; SP, SPARC domain; TY, thyroglobulin domain. (B) Microscale thermophoresis assay for the interaction between fluorescently labeled thrombin and full-length (FL) SMOC1 (FL-SMOC1), SMOC1ΔKazal, (ΔKazal), or the SMOC1 Kazal (Kazal) domain (6 pmol/L-8 nmol/L). The dotted lines indicate the beginning and the end of the laser pulse; n = 4 or 5 independent experiments. (C) Thrombin activity assay in the presence of solvent (Sol; A431 cell supernatant), recombinant SMOC1 (rSMOC1), the overexpressed full-length SMOC1 protein (FL), SMOC1ΔKazal (ΔKazal), and the isolated Kazal domain (Kazal) (all 4 nmol/L); n = 6. ***P < .001, 1-way ANOVA and Bonferroni. (D) Thrombin-induced aggregation of washed human platelets in the presence of Sol (A431 cell supernatant), full-length SMOC1 (FL-SMOC1), the ΔKazal mutant, and the isolated Kazal domain (all 4 nmol/L); n = 5 donors. *P < .05, ***P < .001, 2-way ANOVA and Bonferroni test. (E) Fibrin clot formation (absorbance λ350 nm) in vitro induced by thrombin (0.1 U/mL) in the presence of Sol, full-length SMOC1 (FL), the ΔKazal mutant, and the isolated Kazal domain (all 4 nmol/L); n = 5 independent experiments. ***P < .001, 1-way ANOVA and Bonferroni test.
SMOC1 and thrombin-induced responses in endothelial cells
In addition to affecting hemostasis by activating platelets, thrombin can elicit many other biological effects (for reviews see Popović et al37 and Posma et al38 ). To determine whether SMOC1 also regulates responses to thrombin in cells other that platelets, siRNAs were used to downregulate SMOC1 in human endothelial cells. Thrombin decreased the barrier function of endothelial cells, a phenomenon evidenced by the increased permeability of endothelial cells to fluorescein isothiocyanate–conjugated dextran. The downregulation of SMOC1 significantly attenuated the thrombin-induced increase in permeability, a phenomenon that was not observed following the addition of the recombinant protein (Figure 5A). Consistent with these observations, the thrombin-induced disruption of endothelial CD144-containing cell-cell junctions was potentiated by the addition of recombinant SMOC1 (Figure 5B).
![Effect of SMOC1 on thrombin-induced changes in endothelial cell barrier function. (A) Fluorescein isothiocyanate–labeled dextran (70 kDa; arbitrary units [AU]) permeability in confluent cultures of human endothelial cells treated with a control siRNA (siCTL) or siRNA against SMOC1 (siSMOC1); n = 6 independent cell batches. **P < .01, ***P < .001, 2-way ANOVA and Bonferroni test. (B) CD144, CD31, and DAPI (gray) in endothelial cells under basal conditions (Control) and following the addition of thrombin (0.3 U/mL, 20 minutes). Experiments were performed in the absence and presence of recombinant SMOC1 (rSMOC1). Scale bars, 20 μm. The red boxes denote the area highlighted in the lower images. Comparable results were obtained in 4 additional experiments.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/12/10.1182_blood.2020009405/1/m_bloodbld2020009405f5.png?Expires=1763592109&Signature=Wke-o~ol8wJcTWF8NysW6QZQneOldtzPLwGB2BHK2G4SEGhRaFtpV6ggLwePY3Rkdx4Ex1IQBKF2S7cF0S3kI0VcnyX2hzLFnAfjTy~8f3cYzLWDODU7GyN7nbVTsI3vhriPpPXx5SkWLUX~ZIVLD7Gjl5o63GgbnYlYtLP1lUhMEc9GA~idqVYX8zENROIs1ff123YWPV4h~8I4ehImon6FMC44TIoLZI85PMaddoDk-EidfLSp-WHZsxoPZtvJK-xHTzh2g3BVNrLymTiSz5cOrNdmIKOLoBKv55ohvhOqv~m2vRl2rRBjWcUgqEuge7-vcapQGnrpfng8VVbTPA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Effect of SMOC1 on thrombin-induced changes in endothelial cell barrier function. (A) Fluorescein isothiocyanate–labeled dextran (70 kDa; arbitrary units [AU]) permeability in confluent cultures of human endothelial cells treated with a control siRNA (siCTL) or siRNA against SMOC1 (siSMOC1); n = 6 independent cell batches. **P < .01, ***P < .001, 2-way ANOVA and Bonferroni test. (B) CD144, CD31, and DAPI (gray) in endothelial cells under basal conditions (Control) and following the addition of thrombin (0.3 U/mL, 20 minutes). Experiments were performed in the absence and presence of recombinant SMOC1 (rSMOC1). Scale bars, 20 μm. The red boxes denote the area highlighted in the lower images. Comparable results were obtained in 4 additional experiments.
Effect of SMOC1 on thrombin-induced changes in endothelial cell barrier function. (A) Fluorescein isothiocyanate–labeled dextran (70 kDa; arbitrary units [AU]) permeability in confluent cultures of human endothelial cells treated with a control siRNA (siCTL) or siRNA against SMOC1 (siSMOC1); n = 6 independent cell batches. **P < .01, ***P < .001, 2-way ANOVA and Bonferroni test. (B) CD144, CD31, and DAPI (gray) in endothelial cells under basal conditions (Control) and following the addition of thrombin (0.3 U/mL, 20 minutes). Experiments were performed in the absence and presence of recombinant SMOC1 (rSMOC1). Scale bars, 20 μm. The red boxes denote the area highlighted in the lower images. Comparable results were obtained in 4 additional experiments.
SMOC1 and responses to thrombin in miR-223–deficient murine platelets
Platelet reactivity is altered in diabetes, a phenomenon that has been attributed to changes in multiple signaling pathways.39 Given that miR-223 regulates platelet reactivity,9 as well as the expression of SMOC1,17 the interaction among miR-223, SMOC1, and diabetes was assessed. The expression of SMOC1 was significantly greater in platelets from miR-223–deficient (miR-223y/−) mice than from their WT (miR-223y/+) littermates (Figure 6A), as was that of an additional miR-223 target (ie, β1-integrin). Consistent with the increase in SMOC1 expression, the thrombin-induced phosphorylation of β3-integrin was also increased in miR-223y/− mice (Figure 6B). Thrombin-induced aggregation was also exaggerated in platelets from mice lacking miR-223, which is consistent with a previous study,9 but the response was normalized by the addition of the SMOC1 antibody. The control antibody directed against β-actin was without effect (Figure 6C). In contrast to the situation with human platelets, the SMOC1 antibody failed to affect reactivity to thrombin in samples from WT mice, a phenomenon that may reflect a difference in the affinity of the antibody for the human vs murine SMOC1 protein. Nevertheless, these data suggested that the hyperreactivity of miR-223y/− platelets to thrombin can be attributed, at least in part, to an increase in SMOC1 expression.
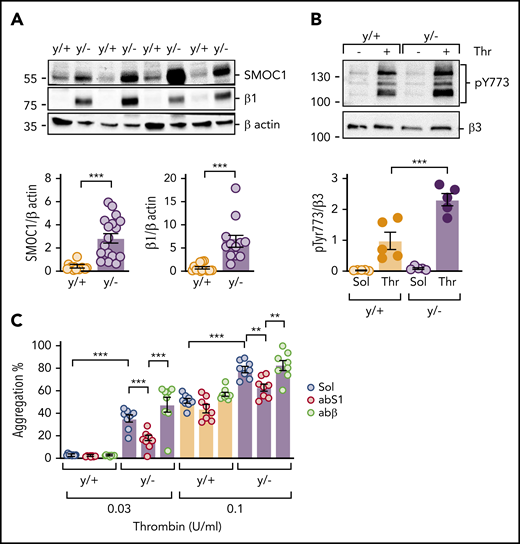
Consequences of miR-223 knockout on platelet SMOC1 expression and responsiveness to thrombin. (A) SMOC1 and β1-integrin (β1) levels in washed platelets from WT (y/+) and miR-223y/− (y/−) mice; n = 14 per group. ***P < .001, Student t test. (B) Thrombin (Thr; 1 U/mL; 10 minutes)–induced phosphorylation of β3-integrin on Tyr773 (pY773) in platelets from WT (y/+) and miR-223y/− (y/−) mice; n = 5 per group. ***P < .001, ANOVA and Bonferroni test. (C) Thrombin-induced aggregation in platelets from WT (y/+) and miR-223y/− (y/−) mice in the presence of solvent (Sol), an antibody against SMOC1 (abS1; 0.625 ng/µL), or an anti–β-actin antibody (abβ; 0.625 ng/µL); n = 8 animals per group. **P < .01, ***P < .001, 2-way ANOVA and Bonferroni test.
Consequences of miR-223 knockout on platelet SMOC1 expression and responsiveness to thrombin. (A) SMOC1 and β1-integrin (β1) levels in washed platelets from WT (y/+) and miR-223y/− (y/−) mice; n = 14 per group. ***P < .001, Student t test. (B) Thrombin (Thr; 1 U/mL; 10 minutes)–induced phosphorylation of β3-integrin on Tyr773 (pY773) in platelets from WT (y/+) and miR-223y/− (y/−) mice; n = 5 per group. ***P < .001, ANOVA and Bonferroni test. (C) Thrombin-induced aggregation in platelets from WT (y/+) and miR-223y/− (y/−) mice in the presence of solvent (Sol), an antibody against SMOC1 (abS1; 0.625 ng/µL), or an anti–β-actin antibody (abβ; 0.625 ng/µL); n = 8 animals per group. **P < .01, ***P < .001, 2-way ANOVA and Bonferroni test.
SMOC1 and responses to thrombin in platelets from diabetic individuals
There are numerous reports of decreased miR-223 levels and hyperreactivity to thrombin in platelets from diabetic individuals.9,10 Therefore, the next step was to determine SMOC1 expression and its potential role in regulating the responsiveness of human platelets to thrombin. Human platelets express PAR1 and PAR4,40 but the hyperresponsiveness to thrombin could not be explained by increased PAR expression, because PAR1 levels tended to be lower in platelets from diabetic individuals vs nondiabetic individuals (Figure 7A). There was also a significant decrease in the expression of the PAR4 receptor in platelets from diabetic individuals. However, diabetes was associated with a significant increase in SMOC1 expression (Figure 7B), as well as β1-integrin expression in human platelets. As expected, platelets from diabetic individuals demonstrated an exaggerated response to low concentrations of thrombin, and consistent with the increase in SMOC1 expression, thrombin-induced aggregation was significantly attenuated by the SMOC1 antibody (Figure 7C).
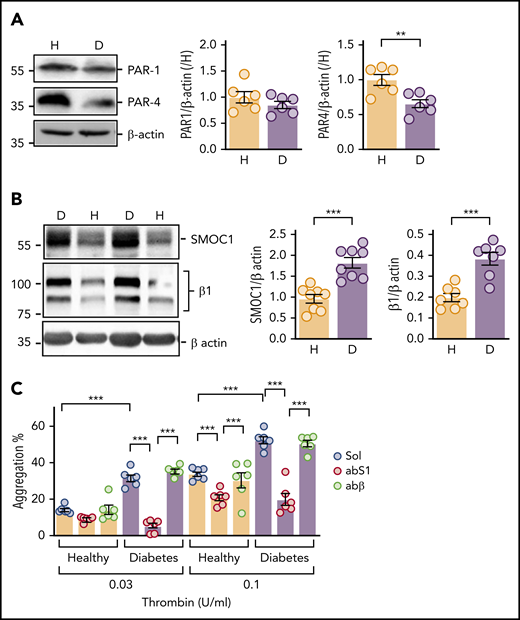
SMOC1 expression in platelets from healthy donors and subjects with type 2 diabetes. Washed platelets were isolated from nondiabetic subjects (healthy; H) and individuals with type 2 diabetes (D). (A) PAR1 and PAR4 expression; n = 6 per group, **P < .01, Student t test. (B) SMOC1 and β1-integrin (β1) levels; n = 8 per group. ***P < .001, Student t test. (C) Thrombin-induced platelet aggregation in the presence of solvent (Sol), an antibody against SMOC1 (abS1), or an anti–β-actin antibody (abβ); n = 6 subjects per group. ***P < .001, 2-way ANOVA and Bonferroni test.
SMOC1 expression in platelets from healthy donors and subjects with type 2 diabetes. Washed platelets were isolated from nondiabetic subjects (healthy; H) and individuals with type 2 diabetes (D). (A) PAR1 and PAR4 expression; n = 6 per group, **P < .01, Student t test. (B) SMOC1 and β1-integrin (β1) levels; n = 8 per group. ***P < .001, Student t test. (C) Thrombin-induced platelet aggregation in the presence of solvent (Sol), an antibody against SMOC1 (abS1), or an anti–β-actin antibody (abβ); n = 6 subjects per group. ***P < .001, 2-way ANOVA and Bonferroni test.
Discussion
The results of this study indicate that the secreted matricellular protein SMOC1 can interact directly with thrombin and increase its activity to potentiate thrombin-induced signaling and aggregation in platelets and alter barrier function in endothelial cells. Not only was the effect apparent under normal physiological conditions, the platelet hyperreactivity to thrombin that was evident in miR-223y/− mice, as well as in humans, with type 2 diabetes, could be attributed to the increased expression of SMOC1. Although SMOC1 is certainly not the only reason for the platelet hyperreactivity associated with type 2 diabetes, it is tempting to speculate that SMOC1 could be used as a biomarker, as well as a potential novel therapeutic target, for the prevention of thrombosis in diabetes.
Not much is known about the physiological role(s) of SMOC1. However, its potential importance is underlined by the fact that altered expression and mutation have been linked with Waardenburg anophthalmia syndrome,19 osteoblast differentiation,16 and some forms of cancer.22,41-43 In the cardiovascular system, SMOC1 has been implicated in hypertension in African Americans,44 and its silencing has been linked with altered responsiveness to angiotensin II,45 as well as defective angiogenesis.17 The results of this study demonstrate a significant role for SMOC1 in the regulation of platelet responsiveness. Indeed, a direct comparison of platelets from WT and SMOC1+/− mice revealed a clear defect in aggregation, platelet-leukocyte aggregate formation and clot retraction, as well as in thrombin-induced Ca2+ signaling and β3-integrin phosphorylation. Importantly, it was possible to restore platelet responsiveness to thrombin in SMOC1-deficient platelets by the addition of recombinant SMOC1. Intriguingly, the hyporesponsiveness of SMOC1-deficient platelets observed in the presence of thrombin was not apparent in platelets stimulated with other platelet agonists. The selectivity to thrombin could not be explained by altered thrombin receptor expression, because the expression of PAR3 and PAR4 was comparable in SMOC1-expressing and -deficient platelets.
Little is known about the mechanisms that regulate the expression of SMOC1. Interestingly, fasting has been linked with increased SMOC1 expression in diabetic wounds and the promotion of wound closure, which may be related to platelet-derived growth factors.46 Hypoxia can increase SMOC1 expression in endothelial cells via the downregulation of miR-223.17 miR-223 was of interest because it is highly expressed in platelets, neutrophils, and monocytes, and it can be transferred to the vascular wall to regulate protein expression and inflammation.47-50 Certainly, SMOC1 expression was higher in platelets from miR-223–deficient mice than from their WT littermates. This is of relevance because the increase in SMOC1 expression coincided with an exaggerated platelet response to thrombin. Platelet miR-223 levels are reported to be decreased in diabetes9,10 ; therefore, SMOC1 levels and responses to thrombin were assessed in washed human platelets from nondiabetic as well as diabetic individuals. This revealed that increased SMOC1 expression was associated with platelet hyperreactivity to thrombin. Because altering SMOC1 expression in isolated human platelets was not possible, experiments were performed using a SMOC1 antibody, which attenuated the thrombin-induced aggregation of platelets from healthy donors and, more importantly, normalized the responsiveness of platelets from individuals with diabetes to thrombin.
The biological actions of SMOC1 have been largely attributed to its ability to bind to TGF-β and bone morphogenetic protein receptors to modulate downstream signaling.15,51 Platelets store large amounts of TGF-β1 that is released at the site of injury following platelet activation3 and acts as a potent chemoattractant for monocytes and fibroblasts, recruiting them to sites of inflammation and repair.52 Less is known about the effects of TGF-β on platelets themselves, but the existence of TGF-β receptors and their downstream signaling mediators, including SMAD2, enables them to respond to TGF-β ligands.53 Reports on the effects of TGF-β1 on platelet function are inconsistent, because TGF-β was found to regulate platelet activity and adenosine diphosphate–induced platelet aggregation through a nontranscriptional signaling pathway.53 However, although TGF-β1–deficient mice exhibited a mild bleeding disorder, as well as faulty platelet aggregation and fibrinogen binding,54 mice with megakaryocyte-specific deletion of TGF-β1 do not show any apparent defect in platelet function in vitro or in vivo.55 Given that SMOC1 was reported to bind to endoglin to antagonize ALK5 signaling in endothelial cells,17 platelet responses to thrombin were studied in the presence of an ALK5 inhibitor. This approach failed to reveal any role for ALK5 in the thrombin-induced phosphorylation of platelet β3-integrin, a response that was almost completely abrogated in the absence of SMOC1.
Thrombin is a serine protease that plays a central role in the coagulation cascade, initiating homeostasis and thrombosis via the cleavage of fibrinogen to fibrin, the activation of platelets, and the conversion of pro-cofactors to active cofactors.56 Given its central role, it is perhaps not surprising that a number of compounds can regulate its activity, the best studied of which is probably hirudin.57 Several endogenous thrombin inhibitors have also been described and are thought to be important in the fine-tuning of thrombin activity. The latter include antithrombin, heparin cofactor II, protein C inhibitor, and the protease nexin 1, which all belong to the serpin family.58 More recently, cartilage oligomeric matrix protein (also known as thrombospondin-5) was added to the list of natural thrombin inhibitors.59 Almost nothing is known about molecules that can enhance thrombin activity. Because our data suggested that SMOC1 selectively potentiated thrombin-induced responses independent of TGF-β receptor activation, we tested the hypothesis that SMOC1 directly interacted with the protease. A microscale thermophoresis assay confirmed the direct association of SMOC1 with thrombin, and it was possible to identify the nonclassical Kazal-type protease inhibitor domain within SMOC1 as essential for this interaction. The Kd for the binding of a commercially available recombinant SMOC-1 to thrombin in vitro was in the micromolar range; although this would be indicative of a low-affinity interaction, the situation may be different in vivo. Indeed, full-length SMOC1, as well as an isolated Kazal domain generated by A431 cells, bound thrombin with a Kd in the nanomolar range. More importantly, an antibody directed against SMOC1 effectively attenuated the thrombin-induced aggregation of platelets from nondiabetic individuals. This indicates a significant interaction between the 2 proteins under normal physiological conditions.
Taking all of our findings together, we can conclude that SMOC1 is a novel activator of thrombin that potentiates its actions in vitro and in different cellular systems. Moreover, plasma levels of SMOC1 may be useful as a biomarker for platelet hyperreactivity associated with diabetes, thus implying that therapeutic approaches directed against circulating SMOC1 could have the potential to normalize platelet responses and prevent the cardiovascular consequences of diabetes. Indeed, it was possible to demonstrate that a peptide corresponding to the Kazal domain of the protein reproduced the effects of the full-length protein. Thus, it will be interesting to determine whether this peptide can affect platelet reactivity and thrombus formation in vivo. During the preparation of this manuscript, SMOC1 was identified as a glucose-responsive hepatokine important for glycemic control.60 In that study, the investigators reported that the circulating (plasma) SMOC1 level correlated with insulin sensitivity and was decreased in obese insulin-resistant humans. These observations contrast with our finding that there was a marked upregulation of SMOC1 levels in platelets from subjects with diabetes. Although this controversy needs to be clarified, the findings confirm that SMOC1 is a protein with a potential regulatory role in the complications associated with type 2 diabetes.
Data sharing requests should be sent to Ingrid Fleming (fleming@em.uni-frankfurt.de).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Isabel Winter, Ürün Ukan, and Oliver Haun for expert technical assistance.
This work was supported by the Deutsche Forschungsgemeinschaft (SFB 834/3 A5; Project ID: 75732319) and the Else Kröner-Fresenius Foundation Research Training Group “Translational Research Innovation - Pharma (TRIP 2.0).”
Authorship
Contribution: F.D.L. designed the research, acquired and analyzed the data, and wrote the manuscript; A.E., A.K., and C.R. acquired data; D.M.z.H. provided essential tools; V.R. and A.W.M. provided the human platelets samples; B.F. and M.S. interpreted data; and I.F. designed the research, analyzed the data, wrote the manuscript, and acquired the funding.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ingrid Fleming, Institute for Vascular Signalling, Centre for Molecular Medicine, Goethe University, Theodor Stern Kai 7, 60596 Frankfurt am Main, Germany; e-mail: fleming@em.uni-frankfurt.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal