

Key Points
Platelet activation increases PKM2 dimer formation, which modulates platelet functions and arterial thrombosis.
PKM2 regulates PI3K-mediated Akt and GSK3 signaling.

Abstract
Very little is known about the role of metabolic regulatory mechanisms in platelet activation and thrombosis. Dimeric pyruvate kinase M2 (PKM2) is a crucial regulator of aerobic glycolysis that facilitates the production of lactate and metabolic reprogramming. Herein, we report that limiting PKM2 dimer formation, using the small molecule inhibitor ML265, negatively regulates lactate production and glucose uptake in human and murine stimulated platelets. Furthermore, limiting PKM2 dimer formation reduced agonist-induced platelet activation, aggregation, clot retraction, and thrombus formation under arterial shear stress in vitro in both human and murine platelets. Mechanistically, limiting PKM2 dimerization downregulated phosphatidylinositol 3-kinase (PI3K)-mediated protein kinase B or serine/threonine-specific protein kinase (Akt)/glycogen synthase kinase 3 (GSK3) signaling in human and murine platelets. To provide further evidence for the role of PKM2 in platelet function, we generated a megakaryocyte or platelet-specific PKM2−/− mutant strain (PKM2fl/flPF4Cre+). Platelet-specific PKM2-deficient mice exhibited impaired agonist-induced platelet activation, aggregation, clot retraction, and PI3K-mediated Akt/GSK3 signaling and were less susceptible to arterial thrombosis in FeCl3 injury–induced carotid- and laser injury–induced mesenteric artery thrombosis models, without altering hemostasis. Wild-type mice treated with ML265 were less susceptible to arterial thrombosis with unaltered tail bleeding times. These findings reveal a major role for PKM2 in coordinating multiple aspects of platelet function, from metabolism to cellular signaling to thrombosis, and implicate PKM2 as a potential target for antithrombotic therapeutic intervention.
Introduction
Acute coronary syndrome and stroke are leading causes of mortality and morbidity and result in immense health and economic burden. Current strategies to prevent acute coronary syndrome and ischemic stroke in at-risk patients rely on antiplatelet drugs (eg, aspirin, P2Y12 inhibitors), which do not translate into clinical efficacy in one-third of patients.1,2 More potent antiplatelet agents such as glycoprotein IIb/IIIa inhibitors (eg, abciximab) are associated with bleeding complications and are not suitable for long-term use. Therefore, understanding the cellular mechanism that regulates platelet activation and thrombosis is of considerable importance.
Recently, metabolic pathways are increasingly being recognized as potential targets for therapeutic interventions. Although bioenergetic profile studies performed in platelets suggest the existence of metabolic plasticity,3 very little is known about the mechanistic role of cellular metabolism in platelet function. Pyruvate kinase (PK) is a key enzyme that catalyzes the final step of glycolysis, which involves an irreversible transphosphorylation of phosphoenolpyruvate to produce pyruvate and adenosine triphosphate (ATP).4 Mammalian genomes express 4 kinds of PK isoforms (PKL, PKR, PKM1, and PKM2). PKL is expressed primarily in liver cells, whereas PKR expression is limited to red blood cells. Most other types of cells express the alternatively spliced products of the PKM gene, either PKM1 and/or PKM2.5 The PKM1 isoform is expressed in most adult tissues that have high catabolic demands such as muscle and brain, whereas PKM2 is primarily expressed in highly proliferative cells such as stem cells, embryonic cells, and tumor cells that are characterized by high anabolic demand.6 Unlike other isoforms of PK that function only as tetramers, PKM2 exists as tetramers and dimers that are composed of the same monomers but with different biological activities.7
Compared with dimeric PKM2, tetrameric PKM2 has more ubiquitous roles. The tetrameric form of PKM2 has a high PK activity and primarily aids the final step of glycolysis; the dimeric form of PKM2 has a low PK activity that provides a switch for energy metabolism by regulating aerobic glycolysis (conversion of glucose to lactate in the presence of oxygen, a phenomenon referred to as the “Warburg effect” in tumor cells).8-10 Aerobic glycolysis is not highly prevalent and is a metabolic choice of certain cell types, depending on energy requirements and physiological situations. Platelets contain both PKM1 and PKM2, but the existence of dimeric and tetrameric forms of PKM2 in platelets, and their physiological significance, is not well understood. One possibility is that PKM2 may contribute to multiple aspects of platelet function by regulating lactate production and glucose uptake. In addition, PKM2 may regulate platelet functions by acting as a protein tyrosine kinase, an activity that is unique to PKM2 rather than PKM1.11-13
ML265 is a small molecule activator that stabilizes PKM2 tetramers while preventing the formation of PKM2 dimers.14,15 In the current study, we report that platelet activation is associated with increased PKM2 dimer formation, whereas limiting the formation of PKM2 dimers by ML265 treatment inhibits agonist-induced human and murine platelet function by downregulating phosphatidylinositol 3-kinase (PI3K)-mediated protein kinase B or serine/threonine-specific protein kinase/glycogen synthase kinase 3 (Akt/GSK3) signaling. Furthermore, genetic deletion of PKM2, specifically in murine platelets, inhibited stimulus-dependent platelet activation and aggregation in vitro and arterial thrombosis in vivo. These findings suggest that limiting PKM2 dimerization could be a potential future target for antithrombotic therapeutic intervention.
Materials and methods
Detailed information on the study materials and methods is available in the supplemental Materials and methods (available on the Blood Web site).
Mice
Platelet-specific PKM2-deficient mice (PKM2fl/flPF4Cre+) were generated by crossing PKM2fl/fl9 with PF4Cre+ mice (supplemental Figure 5). All the mice used in the current study were on the C57BL/6J background, and littermate control mice were used. Mice were genotyped by polymerase chain reaction according to protocols from The Jackson Laboratory and as previously described.9 Mice were kept in standard animal housing conditions with controlled temperature and humidity, and they had ad libitum access to standard chow diet and water. Male and female mice on a C57BL/6J background used in the current study were 3 to 4 weeks (14-16 g) or 8 to10 weeks (22-28 g) old. Platelets for infusion were isolated from 4- to 6-month-old donor mice of the same genotype. The University of Iowa Animal Care and Use Committee approved all experiments. In vivo studies were performed according to the current Animal Research: Reporting of In Vivo Experiment guidelines (https://arriveguidelines.org/).
Human subjects
Human venous blood was drawn from healthy donors in accordance with the Declaration of Helsinki and approved by the Institutional Review Board, University of Iowa. All participants gave written informed consent.
Statistical analysis
Results of statistical analysis (numbers, averages, deviation, and statistical tests) are reported in the figures and corresponding legends and were performed in GraphPad Prism. Data are represented as mean ± standard error (SEM) in all the figures. For statistical analysis, GraphPad Prism software version 8 was used. Shapiro-Wilk tests and Brown-Forsythe tests were used for normality and variance, respectively. The statistical significance was assessed by using either an unpaired Student t test or one-way analysis of variance (ANOVA) followed by Tukey’s or Holm-Šídák’s multiple comparisons test (for normally distributed data) and the Mann-Whitney U test or Kruskal-Wallis test followed by Dunn’s multiple comparisons test (for non-normally distributed data). P < .05 was considered to be statistically significant.
Results
Platelet activation is associated with elevation of PKM2 dimer formation, glucose uptake, and aerobic glycolysis
Using immunoblotting, we first confirmed the presence of PKM2 in human and mouse platelets (Figure 1A). We found that PKM2 exists in tetrameric and dimeric forms. Compared with quiescent platelets, a significant increase in the expression of PKM2 dimers was observed in human platelets stimulated with convulxin or thrombin (Figure 1B). ML265, a small molecule that is known to stabilize PKM2 tetramers and limit PKM2 dimerization,15 decreased PKM2 dimer formation in human platelets stimulated with convulxin or thrombin. Treatment of human resting platelets with ML265 substantially reduced the formation of PKM2 dimers compared with vehicle-treated quiescent platelets (supplemental Figure 1A). Similar results were observed with mouse platelets (supplemental Figure 3A).
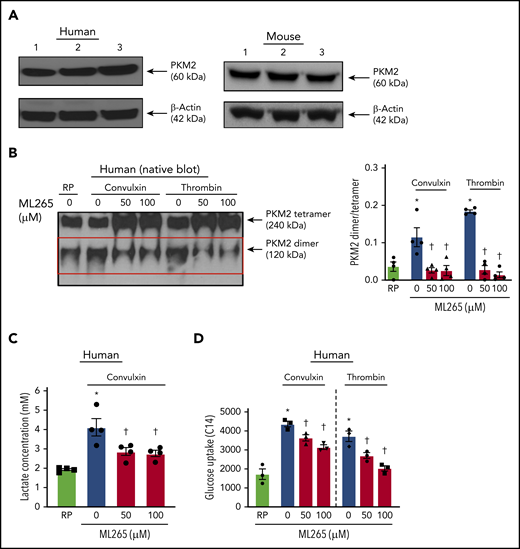
Human platelet activation is associated with an increase in dimeric PKM2 formation. (A) Western blot of PKM2 in human and murine platelets. (B) The left panel shows a representative image of the nonreducing (native) western blot of PKM2 dimer and tetramer formation in human platelets pretreated with vehicle or ML265 and stimulated with convulxin (100 ng/mL) or thrombin (0.1 U/mL). The red box represents PKM2 dimers. The right panel presents the dimer/tetramer ratio densitometry analysis. Values are mean ± SEM, n = 4 individual donors per group. Two-way ANOVA followed by Tukey’s multiple comparisons test. (C) Lactate production in human platelets with convulxin (100 ng/mL). Values are mean ± SEM, n = 4 individual donors per group. One-way ANOVA followed by Tukey’s multiple comparisons test. (D) Effect of PKM2 inhibition on glucose uptake. Values are mean ± SEM, n = 3 individual donors per group. *P < .05 vs resting platelets, †P < .05 vs activated platelets (vehicle). Two-way ANOVA followed by Tukey’s multiple comparisons test. RP, resting platelets.
Human platelet activation is associated with an increase in dimeric PKM2 formation. (A) Western blot of PKM2 in human and murine platelets. (B) The left panel shows a representative image of the nonreducing (native) western blot of PKM2 dimer and tetramer formation in human platelets pretreated with vehicle or ML265 and stimulated with convulxin (100 ng/mL) or thrombin (0.1 U/mL). The red box represents PKM2 dimers. The right panel presents the dimer/tetramer ratio densitometry analysis. Values are mean ± SEM, n = 4 individual donors per group. Two-way ANOVA followed by Tukey’s multiple comparisons test. (C) Lactate production in human platelets with convulxin (100 ng/mL). Values are mean ± SEM, n = 4 individual donors per group. One-way ANOVA followed by Tukey’s multiple comparisons test. (D) Effect of PKM2 inhibition on glucose uptake. Values are mean ± SEM, n = 3 individual donors per group. *P < .05 vs resting platelets, †P < .05 vs activated platelets (vehicle). Two-way ANOVA followed by Tukey’s multiple comparisons test. RP, resting platelets.
Dimeric PKM2 is known to regulate lactate production via aerobic glycolysis. Compared with resting platelets, an approximately twofold rise in lactate production was observed in human platelets stimulated with convulxin, which was significantly decreased in ML265-pretreated groups (Figure 1C). There was no effect on lactate production in resting platelets treated with ML265, compared with vehicle-treated resting samples (supplemental Figure 1B). Glucose uptake is an important step associated with normal platelet activation.16,17 Because the inhibition of dimeric PKM2 downregulated the level of aerobic glycolysis (lactate production), we next sought to determine whether the inhibitory effects of ML265 on platelet functions are solely due to its effects on aerobic glycolysis or if it also regulates glucose uptake. Compared with resting platelets, an approximately twofold increase in glucose uptake was observed in convulxin- or thrombin-stimulated human platelets, which was significantly decreased in ML265-pretreated platelets (Figure 1D).
PKM2 modulates multiple aspects of platelet activation and thrombus formation in vitro
Given the effects of ML265 in regulating glucose uptake and lactate production, we next sought to determine the effects of dimeric and tetrameric PKM2 on platelet functions and thrombosis. Compared with vehicle-treated control, human platelets pretreated with ML265 exhibited reduced stimulus-dependent platelet aggregation against glycoprotein VI (GPVI) agonists (convulxin and collagen) and platelet G protein–coupled receptor (GPCR) agonists (thrombin receptor–activating peptide [TRAP] and adenosine 5′-diphosphate [ADP]) in a concentration-dependent manner (Figure 2A). Phosphatidylserine expression on the platelet surface (a marker of platelet apoptosis) was evaluated by using flow cytometry with annexin V to rule out the possibility that ML265-mediated reduction of platelet aggregation might be an outcome of apoptosis. Treatment with ML265 up to a concentration of 300 µM for 10 minutes did not induce phosphatidylserine surface expression on human platelets (supplemental Figure 2A). Consistent with the inhibition of platelet aggregation, convulxin- and TRAP-induced activation of αIIbβ3 (detected by PAC-1 binding) was reduced in human platelets pretreated with ML265 (Figure 2B). Furthermore, we observed a significant decrease in α-granule (P-selectin exposure) and dense-granule (ATP release) secretion in convulxin- or thrombin-stimulated platelets pretreated with ML265 (Figure 2C-D).
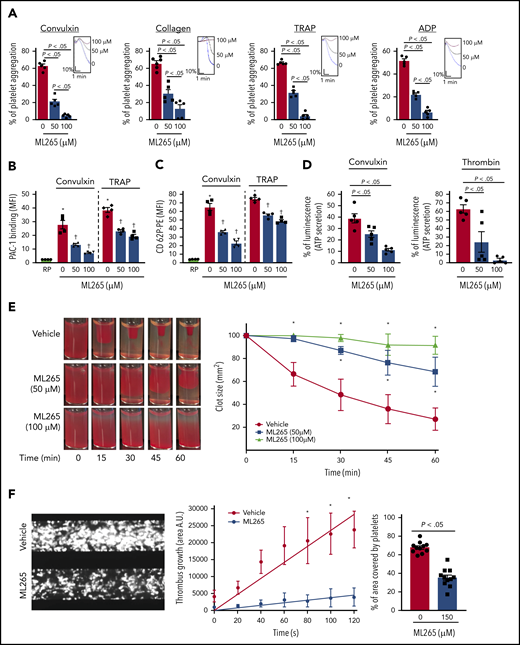
ML265 treatment regulates dimeric PKM2 formation to inhibit multiple aspects of human platelet functions. (A) Human platelet-rich plasma pretreated with vehicle or ML265 and stimulated with convulxin (5 ng/mL), collagen (2.5 μg), TRAP (10 μM), and ADP (5 μM). Results are expressed as the percent change in light transmission with respect to the blank (platelet-poor plasma/buffer without platelets), set at 100%. The upper panel in each bar graph denotes the representative aggregation curves (blue, vehicle-control; black, 50 μM ML265; red, 100 μM ML265). Values are mean ± SEM, n = 5 individual donors per group. One-way ANOVA followed by Tukey’s multiple comparisons test. Effect of dimeric PKM2 inhibition on integrin αIIbβ3 activation (B), P-selectin exposure (C), and ATP secretion (D) from dense granules in stimulated-platelets with convulxin (5 ng/mL), TRAP (50 μM), and thrombin (0.1 U/mL). Values are mean ± SEM, n = 4 to 5 individual donors per group. *P < .05 vs resting platelets, †P < .05 vs vehicle. Two-way ANOVA (B and C) and one-way ANOVA (D) with Tukey’s multiple comparisons test. (E) Clot retraction was measured for 1 hour in platelet-rich plasma, supplemented with red blood cells, after adding 0.25 U/mL of thrombin in the presence of a vehicle or ML265 (50 and 100 μM). The left panels show representative images at different time points; the right panel shows the quantification of clot size with time. Values are mean ± SEM, n = 4 individual donors per group. Two-way ANOVA with Tukey’s multiple comparisons test. (F) Human whole blood pretreated with vehicle or ML265 (150 μM) was perfused over a collagen-coated (100 μg/mL) surface for 5 minutes at a shear rate of 1500 s−1 in a BioFlux Microfluidic flow chamber system from Fluxion Biosciences. The left panel shows the representative image at the end of the assay. The middle panel shows the thrombus growth on the collagen matrix over time. Slopes over time showed that the rate of thrombus growth in ML265-treated whole blood (slope, 37.92) was decreased compared with vehicle control (slope, 237.3). Values are mean ± SEM, n = 3 individual donors per group. *P < .05. Two-way ANOVA with Tukey’s multiple comparisons test. The right panel shows the surface area covered by fluorescent platelets after 5 minutes of perfusion. Three to 4 areas from different areas of the flow chamber were analyzed from each blood sample. Values are mean ± SEM, n = 11 areas per group; Mann-Whitney U test. MFI, mean fluorescence intensity; PE, phycoerythrin.
ML265 treatment regulates dimeric PKM2 formation to inhibit multiple aspects of human platelet functions. (A) Human platelet-rich plasma pretreated with vehicle or ML265 and stimulated with convulxin (5 ng/mL), collagen (2.5 μg), TRAP (10 μM), and ADP (5 μM). Results are expressed as the percent change in light transmission with respect to the blank (platelet-poor plasma/buffer without platelets), set at 100%. The upper panel in each bar graph denotes the representative aggregation curves (blue, vehicle-control; black, 50 μM ML265; red, 100 μM ML265). Values are mean ± SEM, n = 5 individual donors per group. One-way ANOVA followed by Tukey’s multiple comparisons test. Effect of dimeric PKM2 inhibition on integrin αIIbβ3 activation (B), P-selectin exposure (C), and ATP secretion (D) from dense granules in stimulated-platelets with convulxin (5 ng/mL), TRAP (50 μM), and thrombin (0.1 U/mL). Values are mean ± SEM, n = 4 to 5 individual donors per group. *P < .05 vs resting platelets, †P < .05 vs vehicle. Two-way ANOVA (B and C) and one-way ANOVA (D) with Tukey’s multiple comparisons test. (E) Clot retraction was measured for 1 hour in platelet-rich plasma, supplemented with red blood cells, after adding 0.25 U/mL of thrombin in the presence of a vehicle or ML265 (50 and 100 μM). The left panels show representative images at different time points; the right panel shows the quantification of clot size with time. Values are mean ± SEM, n = 4 individual donors per group. Two-way ANOVA with Tukey’s multiple comparisons test. (F) Human whole blood pretreated with vehicle or ML265 (150 μM) was perfused over a collagen-coated (100 μg/mL) surface for 5 minutes at a shear rate of 1500 s−1 in a BioFlux Microfluidic flow chamber system from Fluxion Biosciences. The left panel shows the representative image at the end of the assay. The middle panel shows the thrombus growth on the collagen matrix over time. Slopes over time showed that the rate of thrombus growth in ML265-treated whole blood (slope, 37.92) was decreased compared with vehicle control (slope, 237.3). Values are mean ± SEM, n = 3 individual donors per group. *P < .05. Two-way ANOVA with Tukey’s multiple comparisons test. The right panel shows the surface area covered by fluorescent platelets after 5 minutes of perfusion. Three to 4 areas from different areas of the flow chamber were analyzed from each blood sample. Values are mean ± SEM, n = 11 areas per group; Mann-Whitney U test. MFI, mean fluorescence intensity; PE, phycoerythrin.
We next examined the effects of ML265 on fibrin clot retraction in platelets to ascertain whether ML265 can regulate bidirectional signaling across integrin αIIbβ3 by regulating “outside-in” signaling as well. We observed that clot retraction was significantly inhibited in ML265-treated human platelets that were stimulated with thrombin (Figure 2E), suggesting that dimeric PKM2 regulates integrin αIIbβ3 outside-in signaling. Furthermore, using a microfluidic flow chamber system, we examined whether ML265 can regulate thrombosis ex vivo in the presence of other blood cells. Whole human blood was perfused on a 100 μg/mL collagen-coated surface at an arterial shear rate (1500 s−1) for 5 minutes in the absence or presence of ML265. The size of thrombi was determined by measuring the surface area coverage of fluorescently labeled platelets. ML265 at 50 or 100 µM had no effect compared with vehicle control (not shown). We speculated that a threshold of ML265 may be required to limit PKM2 dimerization in whole blood compared with washed platelets or in platelet-rich plasma. Indeed, at a concentration of 150 µM, a fivefold reduction in the thrombus growth rate along with a decrease in the percentage of the area covered by platelets was observed in the ML265-treated group compared with vehicle control (Figure 2F).
To determine whether dimeric PKM2 also regulates platelet function in mice, we examined the effects of ML265 on multiple aspects of platelet function in murine platelets. Similar to human platelets, ML265 limited PKM2 dimerization and reduced glucose uptake (supplemental Figure 3B-C) in platelets stimulated with convulxin or thrombin. ML265-pretreated platelets also exhibited reduced stimulus-dependent platelet aggregation against GPVI agonists (convulxin and collagen) and GPCR agonists (TRAP and ADP), compared with vehicle control (supplemental Figure 4A). Consistent with the inhibition of platelet aggregation, convulxin- and protease activated receptor 4 (PAR4)-induced activation of αIIbβ3 (detected by JonA binding) was decreased in ML265-pretreated mouse platelets (supplemental Figure 4B). Furthermore, we observed a significant decrease in α-granule secretion (P-selectin exposure), dense-granule secretion (ATP release), and clot retraction (supplemental Figure 4C-E) in stimulus-activated ML265-pretreated platelets.
We next investigated the effects of ML265 on thrombus formation in vitro using a microfluidic flow chamber system. A significant reduction in thrombus growth rate and percentage of the area covered by mouse platelets was observed in the ML265-treated whole blood compared with vehicle control (supplemental Figure 4F). Collectively, these results suggest that PKM2 modulates multiple aspects of platelet function in both human and murine platelets.
Platelet-specific deletion of PKM2 impairs platelet activation
To confirm a mechanistic role for PKM2 in regulating multiple aspects of platelet function and arterial thrombosis, we generated mice that are devoid of PKM2 in megakaryocyte/platelets (PKM2fl/flPF4Cre+) (supplemental Figure 5). Western blotting confirmed the absence of PKM2 in PKM2fl/flPF4Cre+ platelets (Figure 3A) but not in peritoneal macrophages of PKM2fl/flPF4Cre+, suggesting specific deletion of PKM2 from platelets (supplemental Figure 6). The size and morphology of platelets were comparable between PKM2fl/flPF4Cre+ and littermate control PKM2fl/fl mice, as determined by using transmission electron microscopy (Figure 3A). The body weight, complete blood count (supplemental Table 1), and expression levels of platelet surface receptors, including integrin αIIbβ3, glycoprotein Ib, and GPVI, were comparable between PKM2fl/flPF4Cre+ and littermate control PKM2fl/fl mice (supplemental Figure 7). We also looked at the subcellular localization of PKM2 in resting and activated platelets by immunogold staining using transmission electron microscopy. PKM2 was predominantly localized in the α-granules of resting platelets and in the cytoplasm of activated platelets (supplemental Figure 8). Thereafter, we determined the effects of lack of PKM2 on lactate production. Consistent with the effects of ML265 on human platelets, platelet-specific deletion of PKM2 reduced lactate production only in the activated platelets but not in the resting platelets (supplemental Figure 9A). This finding suggests that platelet-specific PKM2-deficient mice could be used as an animal model to further assess the role of PKM2 in regulating multiple aspects of platelet function.
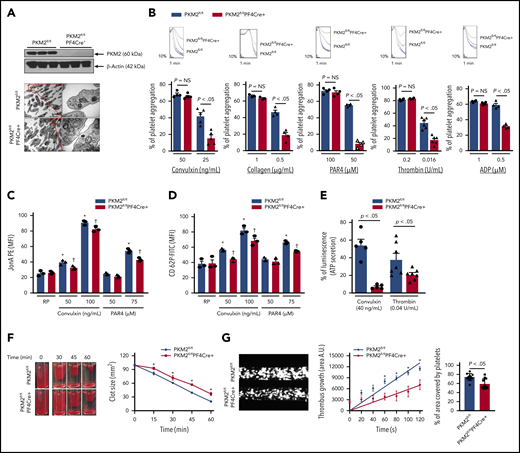
Platelet-specific deletion of PKM2 downregulates a range of platelet functions. (A) The upper panel shows the western blot of PKM2 in platelets from PKM2fl/fl and PKM2fl/flPF4Cre+ mice. The individual lanes are from 3 different mice per group. The lower panel shows the transmission emission microscopy of platelets. The inset in the boxed region is magnified and shown in a microphotograph. Scale bar, 0.5 μm. (B) Platelet-rich plasma or washed platelets from PKM2fl/fl and PKM2fl/flPF4Cre+ were stimulated with different agonists, including convulxin, collagen, PAR4, thrombin, and ADP. Results are expressed as the percent change in light transmission with respect to the blank (platelet-poor plasma/buffer without platelets), set at 100%. The upper panel in each bar graph denotes the representative aggregation curves (blue and red, PKM2fl/fl; black and green, PKM2fl/flPF4Cre+. Values are mean ± SEM, n = 3 to 6 mice per group. *P < .05 vs control. One-way ANOVA followed by Tukey’s multiple comparisons test. Effect of lack of PKM2 on integrin αIIbβ3 activation (C), P-selectin exposure (D), and ATP secretion (E) from dense granules in stimulated platelets with agonists, including convulxin, PAR4, and thrombin. Values are mean ± SEM, n = 3 to 7 mice per group. *P < .05 vs resting platelets, †P < .05 vs PKM2fl/fl. Two-way ANOVA (C and D) and one-way ANOVA (E) followed by Tukey’s multiple comparisons test. (F) Clot retraction was measured for 1 hour in platelet-rich plasma, supplemented with red blood cells, after adding 0.25 U/mL of thrombin. The left panels show the representative images at different time points, and the right panel shows the quantification of the clot size. Platelet-rich plasma was pooled from 5 mice in each group. Values are mean ± SEM, with n = 3 experiments per group. *P < .05 vs PKM2fl/fl, two-way ANOVA with Holm-Šídák’s multiple comparisons test. (G) Mouse whole blood from PKM2fl/fl or PKM2fl/flPF4Cre+ was perfused over a collagen-coated (100 μg/mL) surface for 5 minutes at a shear rate of 1500 s−1 in a BioFlux Microfluidic flow chamber system from Fluxion Biosciences. The left panel shows the representative image at the end of the assay, and the middle panel shows the thrombus growth on the collagen matrix over time. Slopes over time showed that the rate of thrombus growth in the PKM2fl/flPF4Cre+ mice (slope, 58.40) was decreased compared with PKM2fl/fl mice (slope, 108.7). Values are mean ± SEM, n = 3 mice per group. *P < .05. Two-way ANOVA with Tukey’s multiple comparisons test. The right panel shows the surface area covered by fluorescent platelets after 5 minutes of perfusion. Three to 4 areas from different areas of the flow chamber were analyzed from each blood sample; n = 11 areas per group. Mann-Whitney U test. AU, arbitrary unit; FITC, fluorescein isothiocyanate; MFI, mean fluorescence intensity; NS, not significant; PE, phycoerythrin.
Platelet-specific deletion of PKM2 downregulates a range of platelet functions. (A) The upper panel shows the western blot of PKM2 in platelets from PKM2fl/fl and PKM2fl/flPF4Cre+ mice. The individual lanes are from 3 different mice per group. The lower panel shows the transmission emission microscopy of platelets. The inset in the boxed region is magnified and shown in a microphotograph. Scale bar, 0.5 μm. (B) Platelet-rich plasma or washed platelets from PKM2fl/fl and PKM2fl/flPF4Cre+ were stimulated with different agonists, including convulxin, collagen, PAR4, thrombin, and ADP. Results are expressed as the percent change in light transmission with respect to the blank (platelet-poor plasma/buffer without platelets), set at 100%. The upper panel in each bar graph denotes the representative aggregation curves (blue and red, PKM2fl/fl; black and green, PKM2fl/flPF4Cre+. Values are mean ± SEM, n = 3 to 6 mice per group. *P < .05 vs control. One-way ANOVA followed by Tukey’s multiple comparisons test. Effect of lack of PKM2 on integrin αIIbβ3 activation (C), P-selectin exposure (D), and ATP secretion (E) from dense granules in stimulated platelets with agonists, including convulxin, PAR4, and thrombin. Values are mean ± SEM, n = 3 to 7 mice per group. *P < .05 vs resting platelets, †P < .05 vs PKM2fl/fl. Two-way ANOVA (C and D) and one-way ANOVA (E) followed by Tukey’s multiple comparisons test. (F) Clot retraction was measured for 1 hour in platelet-rich plasma, supplemented with red blood cells, after adding 0.25 U/mL of thrombin. The left panels show the representative images at different time points, and the right panel shows the quantification of the clot size. Platelet-rich plasma was pooled from 5 mice in each group. Values are mean ± SEM, with n = 3 experiments per group. *P < .05 vs PKM2fl/fl, two-way ANOVA with Holm-Šídák’s multiple comparisons test. (G) Mouse whole blood from PKM2fl/fl or PKM2fl/flPF4Cre+ was perfused over a collagen-coated (100 μg/mL) surface for 5 minutes at a shear rate of 1500 s−1 in a BioFlux Microfluidic flow chamber system from Fluxion Biosciences. The left panel shows the representative image at the end of the assay, and the middle panel shows the thrombus growth on the collagen matrix over time. Slopes over time showed that the rate of thrombus growth in the PKM2fl/flPF4Cre+ mice (slope, 58.40) was decreased compared with PKM2fl/fl mice (slope, 108.7). Values are mean ± SEM, n = 3 mice per group. *P < .05. Two-way ANOVA with Tukey’s multiple comparisons test. The right panel shows the surface area covered by fluorescent platelets after 5 minutes of perfusion. Three to 4 areas from different areas of the flow chamber were analyzed from each blood sample; n = 11 areas per group. Mann-Whitney U test. AU, arbitrary unit; FITC, fluorescein isothiocyanate; MFI, mean fluorescence intensity; NS, not significant; PE, phycoerythrin.
We next evaluated whether the deletion of PKM2 affects integrin αIIbβ3 “inside-out” and "outside-in" signaling. Platelets from PKM2fl/flPF4Cre+ mice exhibited impaired platelet aggregation upon stimulation with convulxin, collagen, PAR4, thrombin, and ADP (Figure 3B). Consistent with these results, convulxin- and PAR4-induced activation of αIIbβ3 (detected by JONA binding) was decreased in PKM2fl/flPF4Cre+ platelets compared with littermate control PKM2fl/fl platelets (Figure 3C). Furthermore, a significant decrease was observed in α-granule secretion (P-selectin exposure) and dense-granule secretion (ATP release) in PKM2fl/flPF4Cre+ platelets compared with littermate control PKM2fl/fl platelets (Figure 3D-E). The total ATP content was comparable between resting PKM2fl/fl platelets and PKM2fl/flPF4Cre+ platelets (supplemental Figure 9B). Integrin αIIbβ3-mediated clot retraction was also reduced in PKM2fl/flPF4Cre+ platelets compared with control PKM2fl/fl platelets (Figure 3F). On the collagen-coated microfluidic chamber, the thrombus rate and the percentage of the area covered by platelets were significantly decreased in whole blood from PKM2fl/flPF4Cre+ mice compared with control PKM2fl/fl mice (Figure 3G).
To confirm the selectivity of ML265 toward PKM2, its effects were examined in PKM2fl/flPF4Cre+ platelets. Treatment with ML265 significantly reduced convulxin-mediated platelet aggregation in PKM2fl/fl platelets but had no additive inhibitory effect in PKM2fl/flPF4Cre+ platelets (supplemental Figure 10), confirming the specificity of ML265 for PKM2. Collectively, these results provide evidence that at the genetic level PKM2 contributes to platelet activation by regulating diverse aspects of platelet function.
Targeting PKM2 inhibits arterial thrombosis without altering hemostasis in vivo
Having evaluated the effects of PKM2 on platelet activation in vitro, we next evaluated the role of PKM2 in thrombosis and hemostasis in vivo. Male PKΜ2fl/fl and PKM2fl/flPF4Cre+ mice ∼8 weeks old were subjected to FeCl3 injury–induced carotid artery thrombosis. PKM2fl/flPF4Cre+ mice exhibited smaller thrombi and prolonged time to complete occlusion in injured vessels compared with PKM2fl/fl mice (Figure 4A). The prolonged time to occlusion in the PKM2fl/flPF4Cre+ mice was concomitant with a decrease in the rate of thrombus growth. Susceptibility to arterial thrombosis was also evaluated in another model of thrombosis, laser injury–induced mesenteric artery thrombosis, to ensure that the observed effects are applicable in a broader context. The mean fluorescence intensity of the thrombus was significantly decreased in PKM2fl/flPF4Cre+ male mice compared with PKM2fl/fl mice (Figure 4B). Irrespective of the model, these results suggest that PKM2 modulates arterial thrombosis in vivo. Sex-based differences have been reported in rodent models affecting platelet function and thrombosis, in which male mice generally exhibit shorter occlusion time.18 This might be due to the antiplatelet and cardioprotective roles of endogenous estrogens in female mice.19,20 Six- to 8-week-old female PKΜ2fl/fl and PKM2fl/flPF4Cre+ mice were subjected to FeCl3 injury–induced carotid artery thrombosis to assess for sex-based differences. Consistent with previous studies, we also observed sex-based differences in occlusion time in male vs female mice. The injured carotid artery of wild-type (PKM2fl/fl) male mice treated with 5% FeCl3 for 1.5 minutes exhibited complete occlusion in the range of 15 to 18 minutes, whereas similar experimental conditions did not result in occlusion up to 40 minutes in the female mice. To attain comparable levels of occlusion time between male and female wild-type (PKM2fl/fl) mice, we increased the duration of FeCl3 treatment from 1.5 to 2 minutes in female mice. Compared with PKM2fl/fl female mice, PKM2fl/flPF4Cre+ female mice exhibited smaller thrombi and prolonged time to occlusion, concomitant with a decreased thrombus growth rate (Figure 4C). Despite being less susceptible to arterial thrombosis, the tail bleeding time was comparable between PKM2fl/fl and PKM2fl/flPF4Cre+ mice in both male and female mice, suggesting that lack of PKM2 in platelets does not alter hemostasis (Figure 4D).
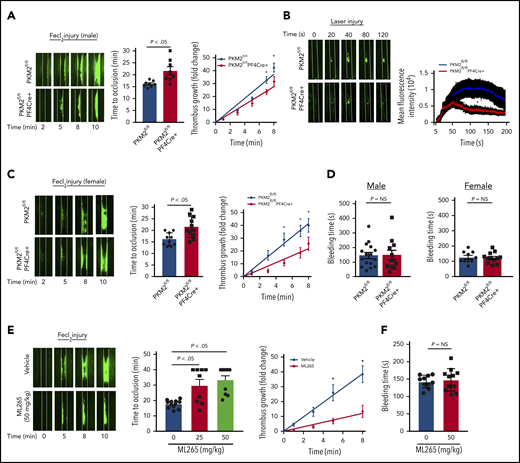
Platelet-specific PKM2-deficient mice were less susceptible to experimental arterial thrombosis. (A) The left panel shows representative microphotographs of carotid artery thrombus (5% FeCl3 injury) as visualized by upright intravital microscopy in male mice. Platelets were labeled ex vivo with calcein green; white lines delineate the arteries. The middle panel shows time to occlusion (n = 8 mice per group). The right panel shows the rate of thrombus growth (n = 8 mice per group). The rate of thrombus growth over a period of 8 minutes was calculated by dividing the area of the thrombus at the time (n) by the area of the same thrombus at the time (0) (defined as the time point at which the thrombus diameter first reached 30 μm). Slopes over time showed that the rate of thrombus growth in the PKM2fl/flPF4Cre+ mice (slope, 3.488) decreased compared with PKM2fl/fl mice (slope, 4.692). *P < .05. Two-way ANOVA with Tukey’s multiple comparisons test. (B) The left panel shows representative microphotographs of mesenteric artery thrombus (laser injury model) as visualized by upright intravital microscopy in male mice. The right panel shows mean fluorescence intensity over time (n = 10-12 vessels from 4 mice per genotype). *P < .05 compared with vehicle control. Mann-Whitney U test. (C) The left panel shows representative microphotographs of carotid artery thrombus (5% FeCl3 injury) as visualized by upright intravital microscopy in female mice. The middle panel shows time to occlusion (n = 10-11 mice per group). The right panel shows the rate of thrombus growth (n = 8 mice per group). Slopes over time showed that the rate of thrombus growth in the PKM2fl/flPF4Cre+ mice (slope, 2.723) decreased compared with PKM2fl/fl mice (slope, 5.175). *P < .05. Two-way ANOVA with Tukey’s multiple comparisons test. (D) Tail bleeding assay in male and female mice. The tail-transection bleeding time was determined as the time taken for the initial cessation of bleeding after transection. Each symbol represents a single mouse; n = 10 to 16 mice per group. Mann-Whitney U test. (E) The left panel shows representative microphotographs of carotid artery thrombus (5% FeCl3 injury) as visualized by upright intravital microscopy in male mice infused with vehicle or ML265. The middle panel shows time to occlusion, and the right panel shows the rate of thrombus growth. Slopes over time showed that the rate of thrombus growth in the ML265-pretreated mice (slope, 1.531) decreased compared with vehicle control (slope, 4.862). Values are expressed as ± SEM. n = 8 to 11 mice per group. *P < .05. Two-way ANOVA with Tukey’s multiple comparisons test. (F) Tail bleeding assay in male mice pretreated with vehicle or ML265. The horizontal bar shows the mean of each group ± SEM, n = 10 to 11 per group. Mann-Whitney U test. NS, not significant.
Platelet-specific PKM2-deficient mice were less susceptible to experimental arterial thrombosis. (A) The left panel shows representative microphotographs of carotid artery thrombus (5% FeCl3 injury) as visualized by upright intravital microscopy in male mice. Platelets were labeled ex vivo with calcein green; white lines delineate the arteries. The middle panel shows time to occlusion (n = 8 mice per group). The right panel shows the rate of thrombus growth (n = 8 mice per group). The rate of thrombus growth over a period of 8 minutes was calculated by dividing the area of the thrombus at the time (n) by the area of the same thrombus at the time (0) (defined as the time point at which the thrombus diameter first reached 30 μm). Slopes over time showed that the rate of thrombus growth in the PKM2fl/flPF4Cre+ mice (slope, 3.488) decreased compared with PKM2fl/fl mice (slope, 4.692). *P < .05. Two-way ANOVA with Tukey’s multiple comparisons test. (B) The left panel shows representative microphotographs of mesenteric artery thrombus (laser injury model) as visualized by upright intravital microscopy in male mice. The right panel shows mean fluorescence intensity over time (n = 10-12 vessels from 4 mice per genotype). *P < .05 compared with vehicle control. Mann-Whitney U test. (C) The left panel shows representative microphotographs of carotid artery thrombus (5% FeCl3 injury) as visualized by upright intravital microscopy in female mice. The middle panel shows time to occlusion (n = 10-11 mice per group). The right panel shows the rate of thrombus growth (n = 8 mice per group). Slopes over time showed that the rate of thrombus growth in the PKM2fl/flPF4Cre+ mice (slope, 2.723) decreased compared with PKM2fl/fl mice (slope, 5.175). *P < .05. Two-way ANOVA with Tukey’s multiple comparisons test. (D) Tail bleeding assay in male and female mice. The tail-transection bleeding time was determined as the time taken for the initial cessation of bleeding after transection. Each symbol represents a single mouse; n = 10 to 16 mice per group. Mann-Whitney U test. (E) The left panel shows representative microphotographs of carotid artery thrombus (5% FeCl3 injury) as visualized by upright intravital microscopy in male mice infused with vehicle or ML265. The middle panel shows time to occlusion, and the right panel shows the rate of thrombus growth. Slopes over time showed that the rate of thrombus growth in the ML265-pretreated mice (slope, 1.531) decreased compared with vehicle control (slope, 4.862). Values are expressed as ± SEM. n = 8 to 11 mice per group. *P < .05. Two-way ANOVA with Tukey’s multiple comparisons test. (F) Tail bleeding assay in male mice pretreated with vehicle or ML265. The horizontal bar shows the mean of each group ± SEM, n = 10 to 11 per group. Mann-Whitney U test. NS, not significant.
After confirming the role of PKM2 in modulating thrombosis in vivo, we sought to determine the inhibitory effects of ML265 on thrombosis in mice. Eight- to 10-week-old male wild-type mice on a C57BL6/J background with or without ML265 pretreatment were subjected to FeCl3-induced carotid artery thrombosis. There was an approximately fourfold reduction in the extent of thrombus growth in ML265-treated mice compared with the vehicle-treated group (Figure 4E). In line with this observation, the time to complete vessel occlusion was significantly prolonged in the mice pretreated with ML265 compared with the vehicle-treated mice. A large number of mice that were pretreated with ML265 did not occlude up to 40 minutes, whereas the time to occlusion for all mice in the control group was ∼20 minutes. Based on the antithrombotic effects of ML265, its impact on hemostasis was evaluated by measuring the tail-transection bleeding time. The mean time to cessation of bleeding was found to be comparable between the ML265- and vehicle-treated groups, suggesting that ML265 treatment does not alter hemostasis (Figure 4F). Together, these results suggest that PKM2 regulates arterial thrombosis in vivo, and targeting dimeric PKM2 with ML265 could be a potential antithrombotic therapeutic intervention.
PKM2 regulates PI3K-mediated Akt/GSK3 signaling in platelets
A previous study by Wang et al21 reported that PKM2 promotes cell migration and inhibits autophagy by mediating PI3K/Akt signaling and contributes to the development of malignant gastric cancer. The PI3K/Akt signaling pathway is also known to be involved in both inside-out and outside-in signaling during platelet activation.22,23 Because pretreatment of human platelets with ML265 downregulated both inside-out and outside-in signaling in stimulated platelets, we further determined whether targeting PKM2 with ML265 attenuates PI3K-mediated signaling in human and murine platelets. Pretreating human platelets with ML265 downregulated convulxin- or thrombin-mediated Akt and GSK3β phosphorylation compared with vehicle control (Figure 5A). In line with these observations, pretreating murine platelets with ML265 also attenuated convulxin- or thrombin-mediated Akt and GSK3β phosphorylation compared with vehicle control (supplemental Figure 11).
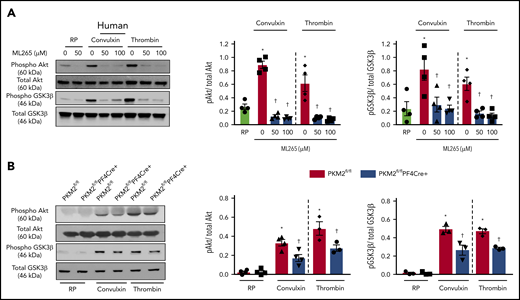
PKM2 regulates PI3K-mediated Akt/GSK3β signaling in platelets. (A) Human and mouse platelets were pretreated with vehicle or ML265 (50 and 100 μM) for 10 minutes at room temperature before stimulation with convulxin (100 ng/mL) or thrombin (0.1 U/ml) for 10 minutes. The left panels show representative western blots for phospho-Akt (Ser 473), total Akt, phospho-GSK3β, and total GSK3β. The middle and right panels show densitometry analysis of immunoblots. Values are mean ± SEM, n = 4 per group. *P < .05 vs resting platelets, †P < .05 vs activated platelets (vehicle). Two-way ANOVA with Tukey’s multiple comparisons test. (B) Platelets from PKM2fl/fl or PKM2fl/flPF4Cre+ were stimulated with convulxin (100 ng/mL) or thrombin (0.1 U/mL) for 10 minutes. The left panels show representative western blots for phospho-Akt (Ser 473), total Akt, phospho-GSK3β, and total GSK3β; the middle and right panels show densitometry analysis of immunoblots. Values are mean ± SEM, n = 3 to 4 per group. *P < .05 vs resting platelets, †P < .05 vs PKM2fl/fl. Two-way ANOVA with Tukey’s multiple comparisons test. RP, resting platelets.
PKM2 regulates PI3K-mediated Akt/GSK3β signaling in platelets. (A) Human and mouse platelets were pretreated with vehicle or ML265 (50 and 100 μM) for 10 minutes at room temperature before stimulation with convulxin (100 ng/mL) or thrombin (0.1 U/ml) for 10 minutes. The left panels show representative western blots for phospho-Akt (Ser 473), total Akt, phospho-GSK3β, and total GSK3β. The middle and right panels show densitometry analysis of immunoblots. Values are mean ± SEM, n = 4 per group. *P < .05 vs resting platelets, †P < .05 vs activated platelets (vehicle). Two-way ANOVA with Tukey’s multiple comparisons test. (B) Platelets from PKM2fl/fl or PKM2fl/flPF4Cre+ were stimulated with convulxin (100 ng/mL) or thrombin (0.1 U/mL) for 10 minutes. The left panels show representative western blots for phospho-Akt (Ser 473), total Akt, phospho-GSK3β, and total GSK3β; the middle and right panels show densitometry analysis of immunoblots. Values are mean ± SEM, n = 3 to 4 per group. *P < .05 vs resting platelets, †P < .05 vs PKM2fl/fl. Two-way ANOVA with Tukey’s multiple comparisons test. RP, resting platelets.
Consistent with inhibitor studies, genetic deletion of PKM2 reduced stimuli-induced phosphorylation levels of both Akt and GSK3β in response to convulxin or thrombin (Figure 5B). PKM2 has protein tyrosine kinase activity and may phosphorylate Akt and GSK3β directly to potentiate platelet activation. However, coimmunoprecipitation studies suggested that PKM2 does not interact with Akt or GSK3β (not shown), arguing against the possibility that PKM2 phosphorylates Akt and GSK3β directly in platelets. Collectively, these results provide evidence that targeting PKM2 directly or indirectly downregulates PI3K-mediated Akt/GSK3β signaling, resulting in decreased platelet activation.
Discussion
The interplay between metabolic pathways and cell signaling contributes to several critical processes, including innate and adaptive immunity, carcinogenesis, stem cell proliferation, and inflammation. Little is known about the role of metabolic regulatory mechanisms in platelet activation and thrombosis. Herein, we found that the glycolytic enzyme PKM2 mechanistically regulates platelet function via PI3K-mediated Akt/GSK3 signaling. Animal studies using platelet-specific PKM2-deficient mice confirmed a role of PKM2 in regulating multiple aspects of platelet function and arterial thrombosis.
Platelet activation is accompanied by an increase in glucose uptake and lactate production.16,17,24,25 The current study found that stimulus-dependent platelet activation induces dimeric PKM2 expression with increased glucose uptake and lactate production, which could be inhibited by using the small molecule inhibitor ML265. A recent study by Kulkarni et al25 also suggested that diarylsulfonamide (DASA-58), a small molecule modulator of PKM2, inhibits aerobic glycolysis and agonist-induced platelet responses. Of note, the study did not determine whether platelet activation is accompanied by an increase in PKM2 dimers. Why is there upregulation of dimeric PKM2 (a driver of aerobic glycolysis) after platelet activation? One possibility is that it may be an adaptive response to fulfill the high-energy demand of activated platelets to rapidly form thrombi. Paradoxically, despite less efficiency, the rate of ATP production is 100 times faster in aerobic glycolysis compared with oxidative phosphorylation.26
Our study also suggests a link between dimeric PKM2-mediated metabolism and platelet function. We found that dimeric PKM2 modulates multiple aspects of platelet function in both human and murine platelets in vitro, including integrin αIIbβ3 signaling, α- and dense-granule secretion, and clot retraction in response to stimulation of GPVI (convulxin and collagen) or GPCR (TRAP and ADP). Furthermore, using whole blood, we found that dimeric PKM2 regulates platelet aggregation/thrombus formation on a collagen matrix under arterial shear stress conditions in vitro. Together, these results suggest that the presence of the dimeric PKM2 isoform in platelets enables platelets to adapt quickly to cellular responses, including integrin activation, aggregation, clot retraction, and thrombus formation. We also found a mechanistic link between PKM2 and PI3K-mediated Akt/GSK3 signaling in platelets. In addition to its role as a metabolic enzyme, a nonmetabolic protein tyrosine kinase activity specific to PKM2 has been implicated in several cell types.11-13 The possibility that PKM2 regulates various platelet functions via its nonmetabolic activity cannot be ruled out. In addition, depending on the stimuli, PKM2 is prone to a range of posttranslational modifications (phosphorylation, acetylation, oxidation, ubiquitination, and sumoylation) that can modulate its structural and functional properties.27 For instance, acetylation of lysine K305 inhibits PKM2 activity and promotes lysosome-dependent degradation of PKM2 via chaperone-mediated autophagy,28 whereas high concentration of reactive oxygen species can promote oxidation of PKM2 at cysteine 358, which can facilitate the formation of a less active dimeric PKM2 form.29 It would therefore be interesting to target these PKM2 posttranslational modifications and investigate their effects on platelets.
To confirm the role of PKM2 in regulating multiple aspects of platelet function in vivo, we generated platelet-specific PKM2-deficient mice. Consistent with the inhibitor studies, genetic ablation of PKM2 inhibited lactate production in stimulated platelets, concomitant with decreased integrin αIIbβ3 activation, P-selectin surface expression, platelet aggregation, and clot retraction against GPVI and GPCR agonists. We also found that the deletion of PKM2 in platelets downregulated PI3K-mediated Akt/GSK3 signaling in platelets. Furthermore, platelet-specific PKM2-deficient mice or wild-type mice pretreated with ML265 were less susceptible to arterial thrombosis in 2 experimental models of arterial thrombosis. Interestingly, we found that the effect of ML265 on arterial thrombosis was more pronounced (50% of mice did not occlude) than the platelet-specific deficiency of PKM2 in mice. We speculate that this action might be due to the inhibitory effect of ML265 on dimeric PKM2 in other cell types in addition to platelets such as neutrophils and monocytes that are known to exhibit a high rate of aerobic glycolysis. Neutrophils and monocytes are also known to contribute to thrombus propagation through several mechanisms involving tissue factor, extracellular nuclear traps, and platelet–leukocyte aggregates.30 Despite affecting thrombosis, the deletion of PKM2 in platelets did not affect tail bleeding time in mice, suggesting a normal hemostatic response. This might be because the inhibitory effect of targeting PKM2 dimers on platelet function was observed at suboptimal doses of agonists, whereas no effect was observed at higher agonist concentrations.
Platelet hyperactivation and thrombotic risk are associated with several pre-comorbid conditions, including obesity, hypertension, hyperlipidemia, atherosclerosis, and cancer. These pre-comorbid conditions often increase the risk of coronary artery disease or stroke. Current antiplatelet therapies are often effective in preventing primary or secondary coronary events in patients at high risk, and safety remains a concern due to a significant risk of bleeding. A particular strength of our study is the observation that therapeutic targeting of PKM2 with ML265 or its genetic deletion inhibited platelet functions in human and murine platelets and experimental thrombosis in mice without affecting hemostasis. This study provides an alternative approach to combat thrombosis by targeting a metabolic enzyme in platelets, in contrast to the conventionally used antiplatelet therapies that include cyclooxygenase inhibitors and platelet receptor antagonists (eg, P2Y12, PAR1, glycoprotein IIb/IIIa).31
Despite its strength, the current study has limitations. It was performed on human platelets that were donated by healthy individuals. Future studies are warranted to see whether targeting dimeric PKM2 would inhibit platelet function isolated from patients with coronary artery disease.
In summary, we have uncovered a novel regulatory pathway in platelets mediated by metabolic enzyme PKM2 that coordinates multiple aspects of platelet function from metabolism to cellular signaling to thrombosis (Figure 6).
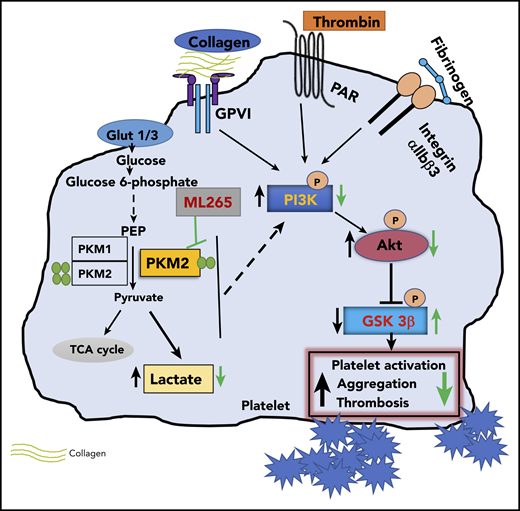
Summary scheme graphic showing a link between PKM2-mediated cellular signaling, platelet function, and thrombosis. In the presence of excess glucose, dimeric PKM2 helps in glucose uptake, and lactate production enhances platelet activation and aggregation via PI3K-mediated Akt/GSK3β signaling to submaximal stimulus-response to GPVI agonists (collagen and convulxin) and GPCR agonists (ADP and thrombin). Glut1, glucose transporter 1; Glut3, glucose transporter 3; PEP, phosphoenolpyruvate; TCA, tricarboxylic acid.
Summary scheme graphic showing a link between PKM2-mediated cellular signaling, platelet function, and thrombosis. In the presence of excess glucose, dimeric PKM2 helps in glucose uptake, and lactate production enhances platelet activation and aggregation via PI3K-mediated Akt/GSK3β signaling to submaximal stimulus-response to GPVI agonists (collagen and convulxin) and GPCR agonists (ADP and thrombin). Glut1, glucose transporter 1; Glut3, glucose transporter 3; PEP, phosphoenolpyruvate; TCA, tricarboxylic acid.
The data that support the findings of this study are available from the corresponding author upon reasonable request (Anil K. Chauhan; e-mail: anil-chauhan@uiowa.edu).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Matthew G. Vander Heiden for sharing the PKM2fl/fl mutant strain.
The Anil K. Chauhan laboratory is supported by grants from the National Institutes of Health, National Heart, Lung, and Blood Institute (R35HL139926) and the National Institute of Neurological Disorders and Stroke (R01NS109910 and U01NS113388) and by an Established Investigator Award (18EIA33900009) from the American Heart Association.
Authorship
Contribution: M.K.N. and A.K.C. were responsible for conceptualization; M.K.N., M.G., G.D.F., and N.D. analyzed the data; M.K.N., M.G., G.D.F., N.D., M.J., K.R.M., and M.J.P. performed investigations; S.R.L. contributed intellectual input; M.K.N., G.D.F., and A.K.C. wrote the original draft; and all authors reviewed and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Anil K. Chauhan, University of Iowa, Department of Internal Medicine, 3120 Medical Labs, Iowa City, IA 52242; e-mail: anil-chauhan@uiowa.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal