

Key Points
Pre-HCT pulmonary viral infection, microbiome depletion, lung inflammation, and epithelial injury identify post-HCT lung injury in children.
Strategies to optimize the pre-HCT pulmonary microbiome and mitigate pulmonary inflammation may improve the safety of pediatric HCT.

Abstract
Lung injury after pediatric allogeneic hematopoietic cell transplantation (HCT) is a common and disastrous complication that threatens long-term survival. To develop strategies to prevent lung injury, novel tools are needed to comprehensively assess lung health in HCT candidates. Therefore, this study analyzed biospecimens from 181 pediatric HCT candidates who underwent routine pre-HCT bronchoalveolar lavage (BAL) at the University Medical Center Utrecht between 2005 and 2016. BAL fluid underwent metatranscriptomic sequencing of microbial and human RNA, and unsupervised clustering and generalized linear models were used to associate microbiome gene expression data with the development of post-HCT lung injury. Microbe-gene correlations were validated using a geographically distinct cohort of 18 pediatric HCT candidates. The cumulative incidence of post-HCT lung injury varied significantly according to 4 pre-HCT pulmonary metatranscriptome clusters, with the highest incidence observed in children with pre-HCT viral enrichment and innate immune activation, as well as in children with profound microbial depletion and concomitant natural killer/T-cell activation (P < .001). In contrast, children with pre-HCT pulmonary metatranscriptomes containing diverse oropharyngeal taxa and lacking inflammation rarely developed post-HCT lung injury. In addition, activation of epithelial-epidermal differentiation, mucus production, and cellular adhesion were associated with fatal post-HCT lung injury. In a separate validation cohort, associations among pulmonary respiratory viral load, oropharyngeal taxa, and pulmonary gene expression were recapitulated; the association with post-HCT lung injury needs to be validated in an independent cohort. This analysis suggests that assessment of the pre-HCT BAL fluid may identify high-risk pediatric HCT candidates who may benefit from pathobiology-targeted interventions.
Introduction
In the United States and Europe, >7500 children undergo hematopoietic cell transplantation (HCT) annually as life-saving treatment of malignancies, immunodeficiencies, hemoglobinopathies, and other disorders.1,2 Transplant survival is limited by the development of pulmonary toxicity, which occurs in 10% to 40% of pediatric HCT recipients and can be attributed to infection, treatment-related toxicity, alloreactivity, and other forms of noninfectious inflammation.3,4 Unfortunately, outcomes for lung-injured pediatric HCT recipients are poor, with ∼40% mortality when mechanical ventilation is required and >60% mortality in the setting of acute respiratory distress syndrome.5-8
Accordingly, it is imperative to identify factors associated with the development of post-HCT lung injury to facilitate prevention, early detection, and biology-targeted treatments. Clinical factors associated with the development of post-HCT lung injury in children include impairment on pre-HCT pulmonary function tests; abnormalities on pre-HCT high-resolution chest tomography (HRCT); the use of myeloablative conditioning and/or specific chemotherapeutic agents, including busulfan, bleomycin, and total body irradiation; and the use of HLA-mismatched or T-cell–replete allografts.3,9-15 A predominant pre-HCT microbiologic factor associated with post-HCT lung injury in children is respiratory virus (RV) infection at the time of transplant conditioning, which may prime the HCT recipient for lung injury, presumably as a result of viral persistence through HCT and/or triggering of alloimmune inflammation as the result of airway epithelial injury.16-20 Other microbiologic factors associated with post-HCT lung injury include pre-HCT cytomegalovirus seropositivity and history of pulmonary bacterial or invasive fungal infection.21-26 Finally, host factors, including elevations in baseline inflammation and tissue injury, may predispose to post-HCT lung injury, although these data are sparse.27-31
To further elucidate pre-HCT biological factors associated with post-HCT lung injury, we applied advances in metatranscriptomic sequencing to a unique cohort of pediatric HCT candidates who underwent routine pre-HCT bronchoalveolar lavage (BAL). Metatranscriptomics combines the benefits of agnostic microbial detection (metagenomic sequencing) with the simultaneous capture of human gene expression.32 Although the pre-HCT pulmonary microbiome has not been previously assessed, numerous aspects of the pre-HCT intestinal microbiome have been strongly associated with post-HCT outcomes. Specifically, loss of intestinal biodiversity and the development of intestinal microbial dominance have been associated with colitis, intestinal graft-versus-host-disease (GVHD), nonrelapse mortality (NRM), and even post-HCT pulmonary complications.33-35 Therefore, we hypothesized that disrupted pre-HCT pulmonary microbiomes paired with gene expression signatures of inflammation and tissue injury would predict the development of post-HCT lung injury in children. Testing these hypotheses is a critical first step in laying the groundwork for targeted detection and intervention strategies for the optimization of pre-HCT lung health and, ultimately, the improvement of HCT survival (Figure 1).
Study design. BAL fluid from pediatric HCT candidates underwent RNA sequencing (RNAseq), and results were correlated with patient outcomes. Bronchoscopy with BAL was performed prior to allogeneic HCT in children. Samples underwent metatranscriptomic RNAseq to identify parallel microbiome and gene expression signatures associated with post-HCT clinical outcomes. These data may inform future strategies to optimize pre-HCT pulmonary health to maximize post-HCT survival in children.
Study design. BAL fluid from pediatric HCT candidates underwent RNA sequencing (RNAseq), and results were correlated with patient outcomes. Bronchoscopy with BAL was performed prior to allogeneic HCT in children. Samples underwent metatranscriptomic RNAseq to identify parallel microbiome and gene expression signatures associated with post-HCT clinical outcomes. These data may inform future strategies to optimize pre-HCT pulmonary health to maximize post-HCT survival in children.
Methods
Study design and patients
This study used a previously described cohort of pediatric allogeneic HCT patients up to the age of 19 years who underwent routine pre-HCT BAL at University Medical Center Utrecht between 2005 and 2016.18 No patients had signs of lower respiratory tract infection at the time of sampling and most patients had no or very mild signs of upper respiratory tract infection. A diagnostic pre-HCT BAL was performed before conditioning in children undergoing general anesthesia for another clinical indication (bone marrow aspirate if malignancy, central line placement if nonmalignant disease, and during HRCT if intubation was required). Most HCT centers do not routinely perform pre-HCT BAL in asymptomatic children or adults, making this a unique cohort of patients. Informed consent was obtained with Institutional Review Board approval (#05/143 and #11/063). All patients subsequently underwent HCT and were followed for ≥2 years. BAL fluid (BALF) testing included RV polymerase chain reaction (PCR); culture for bacteria, yeast, fungi, and Legionella; galactomannan (GM); and cytology (supplemental Methods 1, available on the Blood Web site). When BALF RV testing was positive, HCT was postponed if possible and starting in 2013, post-HCT immunosuppression used for GVHD prophylaxis was extended to prevent alloimmune lung injury. When BALF fungal culture or GM was positive, antifungals and, occasionally, granulocyte transfusions were considered based on HRCT findings and clinical risk. Because patients were asymptomatic, positive bacterial cultures were not treated but were used to guide empiric antibiotics in the event of post-HCT febrile neutropenia. HCT protocols, including conditioning chemotherapy, GVHD and infection prophylaxis, and approach to diagnosis and treatment of post-HCT complications are listed in supplemental Methods 1.
Outcomes
The main outcome was the development of post-HCT lung injury, defined as any new or progressive primary pulmonary disease requiring oxygen therapy for ≥72 hours and associated with ≥1 abnormal lower respiratory tract examination finding (eg, dyspnea, rales, rhonchi) and ≥1 abnormal chest imaging finding (eg, opacity, consolidation, cavitation). This definition was selected to exclude lower acuity and/or transient pulmonary disease. Post-HCT lung injury was then categorized as lower respiratory tract infection because of concern for infection, with or without microbiologic isolation of an organism, or as alloreactive lung syndromes, which were categorized as idiopathic pneumonia syndrome or bronchiolitis obliterans, according to American Thoracic Society and National Institutes of Health chronic GVHD criteria, respectively.36,37 Other outcomes were all-cause mortality, NRM, and fatal lung injury. All outcomes were categorized prior to the conception of this study by 2 unblinded authors who participated in the clinical care of these patients (B.A.V., C.A.L.).
Measurements
Leftover cryopreserved BALF samples were shipped to the University of California, San Francisco and underwent RNA extraction using a previously described protocol (supplemental Methods 2).38,39 Sample RNA was mixed with a spike-in standard, and then sequencing libraries were prepared and underwent 125-nt paired-end sequencing on a NovaSeq 6000 instrument to a target depth of 40 million read-pairs. Resultant .fastq files underwent quality and complexity filtration, followed by alignment to the human genome (hg38). Nonhuman reads were aligned to the NCBI nucleotide database using the IDseq (idseq.net) pipeline v3.13.40 Taxa counts were adjusted for laboratory contamination using a previously described protocol based on the negative association between sample mass and contaminant reads.41 The original mass of each taxa in each sample was backcalculated using the reference spike-in (supplemental Methods 3).39
Data analysis
Because of variations in microbiome composition across patients, microbiome taxa matrices are naturally zero inflated and, thus, were subset to include the most common and most abundant taxa shared across samples.42 Because this excluded many taxa that were uncommon in the cohort but dominant in specific samples (such as certain RV species), we calculated aggregate microbiome traits for each sample, including total bacterial, viral, and fungal mass; α diversity; richness; dominance; and Z-score of the most abundant taxa (supplemental Methods 3). Microbiome summary measures were combined with normalized transformed taxa masses and gene counts, and samples were plotted in 2-dimensional space using the Uniform Manifold Approximation and Projection approach (UMAP).43 Hierarchical clustering was used to group patients and then clinical, microbiome, and gene expression traits were compared among clusters using negative binomial generalized linear models (NB-GLMs; edgeR) and pathway analysis (enrichR).44,45 The cumulative incidence of lung injury, NRM, and fatal lung injury was compared across groups using the Fine and Gray competing risks regressions with subdistribution hazard ratios (SHRs); all-cause mortality was compared across groups using the log-rank test of Kaplan-Meier survival estimates and multivariable Cox proportional-hazards models.46,47 In a separate supervised analysis, we used NB-GLMs to correlate differentially abundant taxa and differentially expressed genes with clinical outcomes.
Validation cohort
Adequately large cohorts of children who have undergone pre-HCT BAL in the absence of clinically significant lower respiratory disease are sparse. Therefore, to validate microbe-gene associations, we prospectively enrolled a cohort of 18 consecutive pediatric allogeneic HCT patients ≤21 years of age who underwent pre-HCT BAL, at the University of California, San Francisco Benioff Children’s Hospital between 2015 and 2020, as evaluation for suspected lower respiratory disease prior to HCT conditioning. Samples were processed as described above, and associations among taxa, genes, and outcomes were tested using gene-taxa correlation matrices with adjustment for false discovery (supplemental Methods 4).
Results
Patient characteristics
During the study interval, 208 of 217 pediatric allogeneic HCT candidates underwent pre-HCT BAL; because of limited biospecimen volume, 192 samples were available for this study. After sequencing, 11 samples were excluded because of low biomass (n = 8) or insufficient human transcriptome coverage (n = 3), leaving a final cohort of 181 patients (Table 1). Samples were obtained a median of 12 days prior to HCT (interquartile range, 9-16). Half of the patients were transplanted for underlying malignancy, allografts were most commonly obtained from umbilical cord blood (65%), and nearly all patients received myeloablative conditioning regimens (95%). The overall incidence of post-HCT lung injury was 21%, and fatal lung injury occurred in 9.4% of patients. On univariate analysis, more recent HCT (2013-2016) was associated with lower rates of lung injury and all-cause, nonrelapse, and lung injury–related mortality (supplemental Table 1). In addition, adolescent patients had higher rates of all-cause mortality and NRM. Nearly half of pre-HCT BALF samples had a pathogen detected in the BAL sample by conventional hospital-based microbiology tests, although this was not associated with the development of post-HCT lung injury or mortality (supplemental Tables 1 and 2). The development of post-HCT adenoviremia was associated with post-HCT lung injury, the development of acute GVHD was associated with overall mortality and NRM, and the development of Epstein-Barr virus reactivation was associated with NRM (supplemental Tables 3 and 4).
Patient characteristics and outcomes
| Characteristic | Incidence, n (%) |
|---|---|
| Age,y | |
| 0 > age < 1 | 22 (12) |
| 1 ≥ age < 5 | 49 (27) |
| 5 ≥ age < 13 | 58 (32) |
| 13 ≥ age < 20 | 52 (29) |
| Sex | |
| Male | 104 (57) |
| Female | 77 (43) |
| Race* | |
| Northern European | 130 (72) |
| African/North African | 13 (7) |
| Multiracial/other | 12 (7) |
| Asian/Southeast Asian | 10 (5.5) |
| Middle Eastern/Persian/Turkish | 10 (5.5) |
| Eastern European/Russian | 6 (3) |
| HCT years | |
| 2005-2008 | 22 (12) |
| 2009-2012 | 66 (37) |
| 2013-2016 | 93 (51) |
| HCT indication | |
| Malignancy | 90 (50) |
| Primary immunodeficiency | 37 (20) |
| Metabolic/inborn error of metabolism | 29 (16) |
| Bone marrow failure syndrome | 21 (12) |
| Autoimmune disease | 4 (2) |
| Allograft donor† | |
| Mismatched UCB | 72 (40) |
| Matched UCB | 45 (25) |
| Matched sibling donor | 37 (20) |
| Matched unrelated donor | 27 (15) |
| Conditioning‡ | |
| Myeloablative | 172 (95) |
| Reduced intensity | 9 (5) |
| Serotherapy | |
| ATG or alemtuzumab | 125 (69) |
| BALF routine microbiology§ | |
| Any abnormality | 90 (50) |
| Virus detected on PCR | 54 (30) |
| Bacterial culture growth | 31 (17) |
| Fungal culture growth | 22 (12) |
| GM positivity | 21 (13) |
| Post-HCT lung injury | |
| Yes | 39 (22) |
| Infection | 13 (7) |
| IPS or unknown | 16 (9) |
| Bronchiolitis obliterans | 10 (6) |
| No | 142 (78) |
| Post-HCT mortality | |
| NRM | 39 (21) |
| Due to lung injury | 17 (9) |
| Not due to lung injury | 22 (12) |
| Relapse mortality | 12 (7) |
| Overall survival | 130 (72) |
| Characteristic | Incidence, n (%) |
|---|---|
| Age,y | |
| 0 > age < 1 | 22 (12) |
| 1 ≥ age < 5 | 49 (27) |
| 5 ≥ age < 13 | 58 (32) |
| 13 ≥ age < 20 | 52 (29) |
| Sex | |
| Male | 104 (57) |
| Female | 77 (43) |
| Race* | |
| Northern European | 130 (72) |
| African/North African | 13 (7) |
| Multiracial/other | 12 (7) |
| Asian/Southeast Asian | 10 (5.5) |
| Middle Eastern/Persian/Turkish | 10 (5.5) |
| Eastern European/Russian | 6 (3) |
| HCT years | |
| 2005-2008 | 22 (12) |
| 2009-2012 | 66 (37) |
| 2013-2016 | 93 (51) |
| HCT indication | |
| Malignancy | 90 (50) |
| Primary immunodeficiency | 37 (20) |
| Metabolic/inborn error of metabolism | 29 (16) |
| Bone marrow failure syndrome | 21 (12) |
| Autoimmune disease | 4 (2) |
| Allograft donor† | |
| Mismatched UCB | 72 (40) |
| Matched UCB | 45 (25) |
| Matched sibling donor | 37 (20) |
| Matched unrelated donor | 27 (15) |
| Conditioning‡ | |
| Myeloablative | 172 (95) |
| Reduced intensity | 9 (5) |
| Serotherapy | |
| ATG or alemtuzumab | 125 (69) |
| BALF routine microbiology§ | |
| Any abnormality | 90 (50) |
| Virus detected on PCR | 54 (30) |
| Bacterial culture growth | 31 (17) |
| Fungal culture growth | 22 (12) |
| GM positivity | 21 (13) |
| Post-HCT lung injury | |
| Yes | 39 (22) |
| Infection | 13 (7) |
| IPS or unknown | 16 (9) |
| Bronchiolitis obliterans | 10 (6) |
| No | 142 (78) |
| Post-HCT mortality | |
| NRM | 39 (21) |
| Due to lung injury | 17 (9) |
| Not due to lung injury | 22 (12) |
| Relapse mortality | 12 (7) |
| Overall survival | 130 (72) |
ATG, antithymocyte globulin; IPS, idiopathic pneumonia syndrome; UCB, umbilical cord blood.
Includes 1 Hispanic patient and 1 patient from Suriname.
The majority of non–UCB grafts were obtained from bone marrow (n = 62); only 2 were from peripheral blood. Includes 1 8/10 mismatched sibling donor and 1 9/10 mismatched unrelated donor. The majority of cytomegalovirus serostatuses were unknown/assumed positive because of the use of UCB.
Most myeloablative conditioning was busulfan/fludarabine (n = 82), busulfan/fludarabine/clofarabine (n = 47), or total body irradiation/etoposide (n = 13). The majority of reduced-intensity conditioning was fludarabine based.
No organisms were detected with other diagnostic tests, such as cytology for Pneumocystis carinii pneumonia or PCR for Mycoplasma or Legionella. Some patients had >1 abnormality.
Unsupervised analysis
We first used an unsupervised approach to assess for combined microbiome-transcriptome networks across patient samples. Because microbiome data are inherently zero inflated with respect to sparse taxa, we represented each sample’s microbiome with a combination of summative microbiome traits and normalized mass of the most abundant taxa within the cohort. These microbiome data were then combined with normalized human protein-coding gene counts and underwent dimensionality reduction using the UMAP approach to yield 4 distinct pre-HCT metatranscriptome clusters (Figure 2A). Spatial overlay of the total RV RNA mass and the total bacterial RNA mass within each BALF sample revealed that the clusters contained significantly different microbes spanning multiple orders of magnitude (Figure 2B-C).
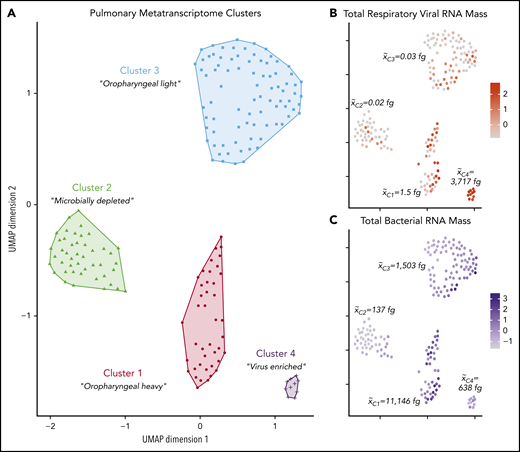
Pre-HCT pulmonary metatranscriptome clusters. Four distinct clusters of BALF samples were identified based on combined microbiome and gene expression data therein. (A) Hierarchical clustering of UMAP coordinates identified 4 groups of patient samples. Centered scaled total respiratory viral RNA mass (B) and total bacterial RNA mass (C) are overlaid on sample coordinates to demonstrate how microbiome characteristics vary across clusters. x̃Cn indicates the median mass of total respiratory viral RNA (B) or total bacteria (C) within each pulmonary metatranscriptome cluster.
Pre-HCT pulmonary metatranscriptome clusters. Four distinct clusters of BALF samples were identified based on combined microbiome and gene expression data therein. (A) Hierarchical clustering of UMAP coordinates identified 4 groups of patient samples. Centered scaled total respiratory viral RNA mass (B) and total bacterial RNA mass (C) are overlaid on sample coordinates to demonstrate how microbiome characteristics vary across clusters. x̃Cn indicates the median mass of total respiratory viral RNA (B) or total bacteria (C) within each pulmonary metatranscriptome cluster.
To further assess metatranscriptomic differences across clusters, we used 4-way analysis of variance–like NB-GLM analysis to identify 84 differentially abundant microbial genera and 442 differentially expressed human genes (false discovery rate [FDR] < 0.1; supplemental Data Set). As shown in Figure 3, cluster 1, which we termed “oropharyngeal (OP) heavy,” was characterized by enrichment of OP commensal taxa, including Streptococcus, Haemophilus, and OP anaerobes, a high degree of microbial richness and diversity, and gene expression suggestive of epithelial cell differentiation. Cluster 2, which we termed “microbially depleted,” was characterized by profound microbiome attenuation with relative sparing of Bacillus and Lactobacillus and enrichment of genes relating to interferon-α and natural killer/T-cell activation (ie, KIRDL4, KLRF1, CTLA4, CCL19, CCR8). Cluster 3, which we termed “OP light,” was characterized by low bacterial biomass with relative sparing of Streptococcus and upregulation of surfactant protein C and hemoglobin synthesis genes. Finally, cluster 4, which we termed “virally enriched,” was characterized by a very high burden of viral RNA; reduced OP commensal taxa; relative sparing of skin taxa, including Staphylococcus, Cutibacterium, and Malassezia; and enrichment of genes relating to acute inflammation and innate immune activation.
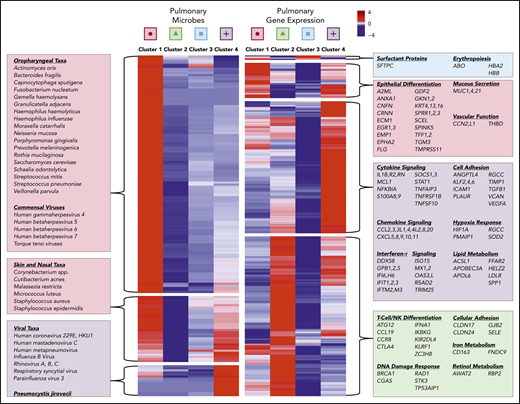
Pre-HCT pulmonary microbiome and gene expression varies by cluster. The microbiome and gene expression characteristics of the 4 BALF clusters varied significantly. The mean centered scaled RNA mass for each taxa (row) in each metatranscriptome cluster (columns) is plotted (left panel). Hierarchical clustering was used to identify 3 breakpoints in the heat map, with clustered taxa identified as OP taxa and commensal viruses, skin and nasal taxa, and viruses. Genes that are differentially expressed across the 4 clusters were identified using 4-way analysis of variance, and the mean variance-stabilizing transformed expression for each gene (row) in each metatranscriptome cluster (column) is plotted (right panel). Hierarchical clustering was used to identify 4 breakpoints in the heat map, with clustered genes enriched for pathways relating to epithelial differentiation, inflammation, cell adhesion, and metabolism.
Pre-HCT pulmonary microbiome and gene expression varies by cluster. The microbiome and gene expression characteristics of the 4 BALF clusters varied significantly. The mean centered scaled RNA mass for each taxa (row) in each metatranscriptome cluster (columns) is plotted (left panel). Hierarchical clustering was used to identify 3 breakpoints in the heat map, with clustered taxa identified as OP taxa and commensal viruses, skin and nasal taxa, and viruses. Genes that are differentially expressed across the 4 clusters were identified using 4-way analysis of variance, and the mean variance-stabilizing transformed expression for each gene (row) in each metatranscriptome cluster (column) is plotted (right panel). Hierarchical clustering was used to identify 4 breakpoints in the heat map, with clustered genes enriched for pathways relating to epithelial differentiation, inflammation, cell adhesion, and metabolism.
To determine the clinical significance of these pre-HCT pulmonary metatranscriptome clusters, we plotted the cumulative incidence of post-HCT lung injury and identified significant differences across the 4 clusters (P < .001; Figure 4A). Relative to patients in the OP-light cluster 3, patients in the microbially depleted cluster 2 had a significantly higher cumulative incidence of lung injury (44.4 ± 0.6% vs 9.5 ± 0.1%; SHR, 5.6; 95% confidence interval [CI], 2.5-12.8; P < .001; Table 2). The cumulative incidence of fatal post-HCT lung injury also varied significantly among metatranscriptome clusters (P = .003, Figure 4B), with patients in the microbially depleted cluster 2 and in the virally enriched cluster 4 showing an SHR of 6.8 (95% CI, 1.9-24.3; P = .003) and 6.3 (95% CI, 1.1-38.2; P = .044), respectively, relative to patients in cluster 3. In a multivariate competing-risks regression accounting for clinical covariates also associated with post-HCT lung injury, metatranscriptome cluster assignment and HCT era remained significantly associated with fatal post-HCT lung injury (P = .032; Table 2). A subanalysis of lung injury subtypes showed that differences in lung injury across metatranscriptome clusters were primarily driven by the development of alloimmune lung injury (P = .002) compared with infectious lung injury (P = .237; supplemental Table 5). Comparison of clinical characteristics across these clusters identified that the OP-heavy cluster 1 had higher rates of bacterial growth and GM positivity, the microbially depleted cluster 2 was more common in adolescents, the OP-light cluster 3 was more common in the most recent epoch, and the virally enriched cluster 4 had higher rates of hospital-based viral PCR positivity (supplemental Table 6).
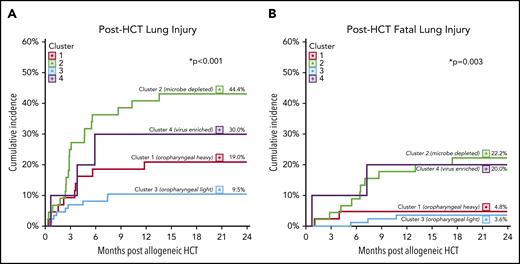
Pre-HCT pulmonary metatranscriptome clusters predict post-HCT clinical outcomes. Pre-HCT BALF metatranscriptome cluster strongly predicted the development of post-HCT lung injury. The cumulative incidences of post-HCT lung injury (A) and fatal post-HCT lung injury (B) were plotted according to the pre-HCT pulmonary metatranscriptome cluster, with death as a competing risk. Significance was assessed using the Fine and Gray competing risks regression.
Pre-HCT pulmonary metatranscriptome clusters predict post-HCT clinical outcomes. Pre-HCT BALF metatranscriptome cluster strongly predicted the development of post-HCT lung injury. The cumulative incidences of post-HCT lung injury (A) and fatal post-HCT lung injury (B) were plotted according to the pre-HCT pulmonary metatranscriptome cluster, with death as a competing risk. Significance was assessed using the Fine and Gray competing risks regression.
Association between pre-HCT pulmonary metatranscriptome and post-HCT clinical outcomes
| Post-HCT outcomes | Pre-HCT pulmonary metatranscriptome | P | |||
|---|---|---|---|---|---|
| Cluster 1 (n = 42) OP-heavy | Cluster 2 (n = 45) Microbially depleted | Cluster 3 (n = 84) OP-light | Cluster 4 (n = 10) Virus enriched | ||
| Lung injury | |||||
| Cumulative incidence, %* | 19.0 ± 0.4 | 44.4 ± 0.6 | 9.5 ± 0.1 | 30.0 ± 2.4 | <.001 |
| SHR (95% CI), univar | 2.1 (0.8-5.7), P = .13 | 5.6 (2.5-12.8), P < .001 | Ref | 3.5 (0.9-13.0), P = .06 | <.001 |
| SHR (95% CI), multivar | 1.8 (0.7-4.8), P = .23 | 3.8 (1.4-9.9), P = .007 | Ref | 2.6 (0.6-10.7), P = .19 | .052 |
| All-cause mortality | |||||
| Kaplan-Meier estimate, %† | 28.6 ± 7.0 | 37.8 ± 7.2 | 25.0 ± 4.7 | 40.0 ± 15.5 | .511 |
| Cox HR, univar | 1.2 (0.6-2.3), P = .70 | 1.5 (0.8-2.9), P = .19 | Ref | 1.7 (0.6-5.1), P = .31 | .518 |
| Cox HR, multivar | 1.0 (0.5-2.1), P = .90 | 1.4 (0.7-2.7), P = .29 | Ref | 1.5 (0.5-4.3), P = .89 | .685 |
| NRM | |||||
| Cumulative incidence, %* | 16.7 ± 0.3 | 31.1 ± 0.5 | 17.9 ± 0.2 | 30.0 ± 2.4 | .312 |
| SHR (95% CI), univar | 0.9 (0.4-2.3), P = .85 | 1.8 (0.9-3.6), P = .12 | Ref | 1.8 (0.5-6.3), P = .36 | .310 |
| SHR (95% CI), multivar | 0.8 (0.3-2.1), P = .69 | 1.6 (0.8-3.3), P = .17 | Ref | 1.5 (0.4-4.9), P = .53 | .360 |
| Fatal lung injury | |||||
| Cumulative incidence, %* | 4.8 ± 0.1 | 22.2 ± 0.4 | 3.6 ± 0.1 | 20.0 ± 1.8 | .003 |
| SHR (95% CI), univar | 1.4 (0.2-8.3), P = .73 | 6.8 (1.9-24.3), P = .003 | Ref | 6.3 (1.1-38.2), P = .044 | .010 |
| SHR (95% CI), multivar | 1.2 (0.2-6.9), P = .87 | 5.3 (1.5-19.0), P = .01 | Ref | 4.4 (0.8-26.1), P = .10 | .032 |
| Post-HCT outcomes | Pre-HCT pulmonary metatranscriptome | P | |||
|---|---|---|---|---|---|
| Cluster 1 (n = 42) OP-heavy | Cluster 2 (n = 45) Microbially depleted | Cluster 3 (n = 84) OP-light | Cluster 4 (n = 10) Virus enriched | ||
| Lung injury | |||||
| Cumulative incidence, %* | 19.0 ± 0.4 | 44.4 ± 0.6 | 9.5 ± 0.1 | 30.0 ± 2.4 | <.001 |
| SHR (95% CI), univar | 2.1 (0.8-5.7), P = .13 | 5.6 (2.5-12.8), P < .001 | Ref | 3.5 (0.9-13.0), P = .06 | <.001 |
| SHR (95% CI), multivar | 1.8 (0.7-4.8), P = .23 | 3.8 (1.4-9.9), P = .007 | Ref | 2.6 (0.6-10.7), P = .19 | .052 |
| All-cause mortality | |||||
| Kaplan-Meier estimate, %† | 28.6 ± 7.0 | 37.8 ± 7.2 | 25.0 ± 4.7 | 40.0 ± 15.5 | .511 |
| Cox HR, univar | 1.2 (0.6-2.3), P = .70 | 1.5 (0.8-2.9), P = .19 | Ref | 1.7 (0.6-5.1), P = .31 | .518 |
| Cox HR, multivar | 1.0 (0.5-2.1), P = .90 | 1.4 (0.7-2.7), P = .29 | Ref | 1.5 (0.5-4.3), P = .89 | .685 |
| NRM | |||||
| Cumulative incidence, %* | 16.7 ± 0.3 | 31.1 ± 0.5 | 17.9 ± 0.2 | 30.0 ± 2.4 | .312 |
| SHR (95% CI), univar | 0.9 (0.4-2.3), P = .85 | 1.8 (0.9-3.6), P = .12 | Ref | 1.8 (0.5-6.3), P = .36 | .310 |
| SHR (95% CI), multivar | 0.8 (0.3-2.1), P = .69 | 1.6 (0.8-3.3), P = .17 | Ref | 1.5 (0.4-4.9), P = .53 | .360 |
| Fatal lung injury | |||||
| Cumulative incidence, %* | 4.8 ± 0.1 | 22.2 ± 0.4 | 3.6 ± 0.1 | 20.0 ± 1.8 | .003 |
| SHR (95% CI), univar | 1.4 (0.2-8.3), P = .73 | 6.8 (1.9-24.3), P = .003 | Ref | 6.3 (1.1-38.2), P = .044 | .010 |
| SHR (95% CI), multivar | 1.2 (0.2-6.9), P = .87 | 5.3 (1.5-19.0), P = .01 | Ref | 4.4 (0.8-26.1), P = .10 | .032 |
Cumulative incidence and variance were calculated using the Fine and Gray cumulative incidence function and compared with the Fine and Gray competing risks regression. Competing risks for lung injury, NRM, and fatal lung injury were death, relapse, and death not related to lung injury, respectively. SHR was calculated with the Fine and Gray competing risks regression. All-cause mortality and standard error were estimated with Kaplan-Meier function and compared using the log-rank test. All-cause mortality hazard ratios were calculated using Cox regression. All multivariate models were adjusted for HCT era. Cluster 3 is the reference group for all comparisons. Bold data indicate statistically significant comparisons.
Multivar, multivariate; Ref, reference; univar, univariate.
Data reported as cumulative incidence estimate with variance.
Estimate is reported as Kaplan-Meier estimate with standard error.
Supervised analysis
Dimensionality-reduction techniques, such as those performed above, are useful to identify patterns, but they do not test associations between outcomes and individual taxa or genes. Therefore, we next performed supervised analysis to identify differentially abundant taxa among patients who developed post-HCT lung injury. Using NB-GLMs, we identified the phyla Negarnaviricota (containing respiratory syncytial virus [RSV]) and Tenericutes (containing Mycoplasma), the genera Enterococcus, Bacillus, and Lactobacillus, and numerous species of Actinomyces, Capnocytophaga, and Porphyromonas to be differentially abundant in pre-HCT BALF from patients who developed post-HCT lung injury (supplemental Figure 1; supplemental Data). These samples were particularly depleted of the OP genera Bacteroides, Fusobacterium, Gemella, Neisseria, and Streptococcus. We noted a virus-specific relationship whereby the development of post-HCT lung injury was positively associated with pre-HCT levels of RSV RNA and negatively associated with pre-HCT levels of rhinovirus RNA (supplemental Figure 2). Of note, rhinoviruses were detected almost exclusively in the clinically low-risk microbially replete clusters 1 (OP heavy) and 3 (OP light).
Using differential gene expression analysis, we identified 165 genes associated with the development of post-HCT lung injury (FDR < 0.1; supplemental Figure 2). Gene ontology analysis demonstrated enrichment for epithelial cell differentiation toward an epidermal phenotype of cornification, keratinization, barrier formation, and peptide cross-linking (adjusted P < .001; supplemental Data Set). We also identified differential expression of genes related to inflammation (ie, CCL19, CCR8, CTLA-4, IL17B, IL26, IKBKG), mucus production (MUC1, MUC21, MUCL3), and cellular adhesion (SELE) in the lungs of patients who later developed post-HCT lung injury. A partially overlapping list of 228 genes was associated with the development of fatal post-HCT lung injury and included numerous genes associated with inflammation (BIRC2, CCL19, IKBKG, IL1RAB, KIRDL4, KLRF1). Expression of niacin receptors 1 and 2 (HCAR2, HCAR3) and hemoglobin synthesis genes (AHSP, HBA2, HBB) were strongly protective against fatal post-HCT lung injury.
Validation cohort
To confirm the association between pulmonary microbes and pulmonary gene expression in an external cohort, we enrolled 18 pediatric pre-HCT patients who underwent pre-HCT BAL (supplemental Table 7). The recent timing of transplantation precluded meaningful analysis of post-HCT clinical outcomes; therefore, we performed metatranscriptomic sequencing as described above and tested for microbe-gene coexpression networks using correlation matrices (supplemental Data). Similar to the Utrecht cohort, we identified strong associations between viral RNA mass and expression of inflammatory genes, such as CCL3L1, CCL4L2, CXCL9, IFNA6, IL1RN, IRF1, TIMP1, and TNFRSF1B (Spearman ρ = 0.54-0.75; FDR P < .1; supplemental Table 8A). We also identified strong associations between OP taxa (ie, Haemophilus, Streptococcus) and epithelial differentiation genes, such as CRNN, ECM1, EMP1, GKN1/2, KRT4/13/16, TFF1/2, and TGM3 (Spearman ρ = 0.54-0.83; FDR P < .1; supplemental Table 8B). Again, we identified an inverse association between hemoglobin synthesis genes (HBA2, HBB) and numerous markers of inflammation and epithelial barrier function (CCL3, CCL4, ECM1, LILRA5, MUC21, TGM3, TIMP1; Spearman ρ = −0.54 to −0.67; FDR P < .1).
Discussion
The overall finding of this study is that pre-HCT pulmonary microbiome-gene expression signatures are predictive of post-HCT lung injury and mortality in pediatric HCT patients. In particular, patients with pulmonary signatures of viral infection, bacterial depletion, immune activation, and epithelial differentiation showed significantly worse outcomes post-HCT. These results add to the current knowledge regarding pre-HCT predictors of HCT-associated lung injury, suggest a biologic mechanism by which the pulmonary microenvironment may prime pediatric patients for post-HCT lung injury, and support a potentially useful diagnostic tool prior to allogeneic HCT.
First, our finding that virus-dominated pre-HCT pulmonary microbiomes were associated with poor outcomes after HCT is novel in a clinically asymptomatic population lacking lower respiratory tract disease. Potential mechanisms may include viral persistence through transplant leading to post-HCT viral pneumonia, viral injury leading to tissue antigen presentation and alloimmune lung syndromes, or virus-induced bacterial dysbiosis predisposing to bacterial infection post-HCT.16-19 Our detection of gene expression enrichment for abnormal cell adhesion and response to hypoxia in virally enriched patients supports the possibility that, despite being asymptomatic, virus-triggered cellular injury was present. In addition, we note the significantly depressed levels of bacterial RNA in the virally enriched cluster 4, a trait that has been associated with predisposition to bacterial dominance and infection in the gut, lung, and oropharynx.33,48
Importantly, the association between pre-HCT RV infection and post-HCT lung injury may be virus specific. Although RSV, parainfluenza virus, and other highly pathogenic viruses were common in the inflamed and lung-injury prone cluster 4, rhinoviruses were almost exclusively found in conjunction with bacterially diverse microbiomes (clusters 1 and 3) and were rarely implicated in post-HCT lung injury. This ecologic finding of viral-bacterial interdependence is in concordance with the growing literature demonstrating a bidirectional cross talk by which RVs can induce bacterial dysbiosis in the airway and certain bacterial species can alter the host responsiveness to viral infection.49 Although hospital-based RV PCR and research-based RNA sequencing (RNAseq) tend to be concordant for viral detection (at least for those species included on viral PCR panels), hospital-based bacterial culture lacks the advantage of describing the entire microbiome community structure. Assessment of the overall microbiome structure in the context of each RV appears particularly important to note given that RVs were detected in all metatranscriptome clusters (by clinical PCR and research-based sequencing). Our research highlights the need for novel supportive care and antiviral therapeutic approaches to mitigate post-HCT lung injury in patients with pre-HCT RV positivity.50,51 The practice of postponing HCT when pre-HCT viral detection is positive has shown encouraging results in select candidates not adversely affected by delayed transplant, but novel approaches are needed for patients who may not have adequate time or immunologic capacity to clear their RV before HCT.52-54
A second finding of this study is that depletion of the pre-HCT pulmonary bacterial microbiome is associated with significantly greater rates of post-HCT lung injury. This finding has not previously been demonstrated in pulmonary studies but is of striking similarity to intestinal microbiome studies associating the loss of intestinal biodiversity with colitis, intestinal GVHD, and NRM.33,34 Intestinal microbiome studies implicate bacterial dysbiosis in the overgrowth of pathogenic taxa, intestinal translocation, and tissue injury leading to the development of infection and alloreactivity.35 Similar mechanisms could explain the association between pulmonary bacterial dysbiosis and adverse outcomes. We hypothesize that decreased pulmonary bacterial load may result from virus-triggered inflammation leading to suppression of bacterial growth, or it may occur as a result of recurrent broad-spectrum antibiotic exposure, which is common throughout the treatment course of patients with hematologic malignancies or primary immunodeficiencies. In support of the latter hypothesis, Ogimi et al showed that antibiotic exposure after HCT is associated with viral progression from upper to lower respiratory tract disease.55 Although patients with malignancy and primary immunodeficiencies tend to have greater antimicrobial use than other patients, we did not detect a greater incidence of the bacterial-deplete cluster 2 in these patient groups, which may suggest that additional factors influence the development of this microbiome characteristic. Current microbiome-modulating strategies under development for intestinal dysbiosis include prebiotics, probiotics, enteral metabolites, and microbiota transplant.35 Ultimately, it remains a challenge how best to restore healthy pulmonary bacterial communities, although cessation of antibiotics and time may be the most efficacious treatments currently available.
A third finding of our work is that pre-HCT pulmonary inflammation is strongly associated with fatal and nonfatal post-HCT lung injury. Although it is not surprising that innate immune activation and cytokine signaling were significantly upregulated in virally enriched pediatric patients, we note that some patients with virus detection by clinical and/or metatranscriptomics testing did not show high levels of antiviral response; this suggests that the paired microbiome-gene expression approach may be a useful method to distinguish active viral infection from asymptomatic or resolving infection. Interestingly, the immune-related gene expression most associated with post-HCT outcomes was dominated by T-cell and natural kill cell–specific markers, such as CTLA4, IL17, and KIRDL4, and co-occurred with pulmonary microbial depletion. Whether this metatranscriptomic signature represents sequelae of distant viral infection or some other injury remains unclear. Possible mechanisms by which this signature might predispose to post-HCT lung injury include predisposition to alloreactivity and/or the inability to control microbial overgrowth; they should be investigated in future studies.
A fourth finding of this study is that activation of pulmonary epithelial-epidermal differentiation is associated with pulmonary microbiomes and post-HCT lung injury. Activation of the epidermal differentiation complex (EDC) is triggered by inflammation, reactive oxygen species, trauma, and cellular injury and has been detected in chronic rhinitis and asthma; however, to our knowledge, this is the first report of EDC activation in the lungs of HCT candidates.56-62 In the short term, EDC activation results in strengthened epithelial integrity, improved barrier function, and antimicrobial protection; therefore, it is likely that EDC activation is a response to injury rather than a trigger of injury. The EDC can be induced in cultured airway epithelial cells using azithromycin and is associated with cornification, decreased cell-cell permeability, and increased tight junction expression.63 In addition to antimicrobial and anti-inflammatory properties, this may partially explain the clinical utility of azithromycin in nonmalignant pulmonary diseases like cystic fibrosis and idiopathic pulmonary fibrosis.64 However, chronic EDC activation can result in fibrosis, metaplasia, and squamous carcinogenesis. In the HCT population, long-term post-HCT azithromycin use has recently been associated with worse airflow decline-free survival and an increased risk for post-HCT hematologic malignancy relapse and secondary neoplasia, although it is unclear whether these findings were related to EDC activation or an off-target biological effect of azithromycin.65-67 Ultimately, future studies are needed to determine whether the EDC can be selectively manipulated prior to HCT to amplify an adaptive response to chronic epithelial damage and prevent post-HCT lung injury.
Our approach benefits from several strengths. First, unlike 16S and 28S/ITS amplicon-based sequencing, metatranscriptomic RNAseq includes bacteria, viruses, and fungi. Second, we used a reference spike-in (External RNA Controls Consortium) to calculate pre-PCR microbe mass, which facilitated calculation of absolute mass changes rather than just relative trends. Third, we used a complex approach to control for laboratory contamination. Fourth, we used unsupervised analysis and confirmed our findings with a supervised approach. Fifth, we combined microbiome measurement with human gene expression to increase biological relevance.
A first limitation of this work is that few centers perform routine pre-HCT BALs in asymptomatic pediatric patients and bank samples for later research. Although we were able to validate many of the microbe-gene relationships in an external cohort, further external validation is required to corroborate relationships between the pre-HCT pulmonary metatranscriptome and clinical outcomes. If these results can be validated externally, then the next step toward using this information to help pediatric patients might be a clinical trial in which pre-HCT BAL is used to select high-risk patients to receive a specific intervention that is studied relative to placebo or standard-of-care. A second limitation is that this study does not attempt to address the full complexities of pre-HCT factors that may influence the development of each patient’s measured pulmonary metatranscriptome. Factors such as detailed infection history, antibiotic type, dose, and duration, and specific chemotherapeutic regimens could all influence the pulmonary metatranscriptome and should be investigated in future studies. Further mechanistic work is necessary to understand whether the pulmonary metatranscriptome is simply a marker of these exposures or represents a modifiable mediator of post-HCT lung injury. A third limitation is the definition of post-HCT lung injury, which excluded lower-acuity disease and may not have been able to fully differentiate between post-HCT infectious vs noninfectious lung injury.68 A fourth limitation is that, although treating physicians were blinded to BALF sequencing results, they were not blinded to standard clinical microbiology results; these data were used to alter care, which may have blunted an association between standard microbiologic results and post-HCT lung injury and could be responsible for improving outcomes over time.
In summary, metatranscriptomic sequencing of pre-HCT pediatric BALF identified distinct microbiome patterns of viral infection and commensal bacterial depletion linked with innate and adaptive immune activation and response to cellular injury. Pediatric patients with these metatranscriptomic signatures had significantly increased post-HCT lung injury and fatal lung injury. This study is the first to associate the pre-HCT pulmonary metatranscriptome with post-HCT outcomes and invites the possibility for a future precision medicine approach to evaluate pulmonary health prior to HCT. This study also highlights the need to investigate the underlying mechanisms by which these biologic states may predispose to post-HCT lung injury.
Sequencing files and deidentified phenotypic data are available with investigator support at the database of Genotypes and Phenotypes under Study Accession phs002208.v1.p1.
Data sharing requests should be sent to Matt S. Zinter (matt.zinter@ucsf.edu).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by the National Marrow Donor Program Amy Strelzer Manasevit Grant (M.S.Z.), National Institutes of Health, Eunice Kennedy Shriver National Institute of Child Health and Human Development grant K12HD000850 (M.S.Z.), National Institutes of Health, National Heart, Lung, and Blood Institute grants K23HL146936 (M.S.Z.) and R01HL134828 and R35HL140026 (M.A.M.), an American Thoracic Society Foundation Grant (M.S.Z.), and the Chan Zuckerberg Biohub (J.L.D.).
Authorship
Contribution: M.S.Z., C.A.L., C.C.D., J.J.B., and J.L.D. conceived and designed the study; M.S.Z., C.A.L., B.A.V., M.Y.M., C.C.D., J.J.B., and J.L.D. acquired data; M.S.Z., C.A.L., C.C.D., J.J.B., and J.L.D. wrote the manuscript; M.S.Z., M.Y.M., M.S., C.C.D., J.J.B., and J.L.D. performed statistical analyses; M.S.Z., C.A.L., B.A.V., M.Y.M., S.S., G.R., C.C.D., J.J.B., and J.L.D. supervised the study; M.S.Z., C.A.L., C.C.D., J.J.B., and J.L.D. provided administrative, technical, or material support; and all authors analyzed and interpreted data, critically revised the manuscript for important intellectual content, and approved the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Matt S. Zinter, University of California, San Francisco, 550 16th St, San Francisco, CA 94143; e-mail: matt.zinter@ucsf.edu.
