

Key Points
GCK activity is critical in MM harboring RAS mutations.
GCK inhibition induces degradation of IKZF1/3 in a Cereblon-independent fashion.

Abstract
In multiple myeloma (MM), frequent mutations of NRAS, KRAS, or BRAF are found in up to 50% of newly diagnosed patients. The majority of the NRAS, KRAS, and BRAF mutations occur in hotspots causing constitutive activation of the corresponding proteins. Thus, targeting RAS mutation in MM will increase therapeutic efficiency and potentially overcome drug resistance. We identified germinal center kinase (GCK) as a novel therapeutic target in MM with RAS mutation. GCK knockdown (KD) in MM cells demonstrated in vitro and in vivo that silencing of GCK induces MM cell growth inhibition, associated with blocked MKK4/7-JNK phosphorylation and impaired degradation of IKZF1/3, BCL-6, and c-MYC. These effects were rescued by overexpression of a short hairpin RNA (shRNA)-resistant GCK, thereby excluding the potential off-target effects of GCK KD. In contrast, overexpression of shRNA-resistant GCK kinase-dead mutant (K45A) inhibited MM cell proliferation and failed to rescue the effects of GCK KD on MM growth inhibition, indicating that GCK kinase activity is critical for regulating MM cell proliferation and survival. Importantly, the higher sensitivity to GCK KD in RASMut cells suggests that targeting GCK is effective in MM, which harbors RAS mutations. In accordance with the effects of GCK KD, the GCK inhibitor TL4-12 dose-dependently downregulated IKZF1 and BCL-6 and led to MM cell proliferation inhibition accompanied by induction of apoptosis. Here, our data identify GCK as a novel target in RASMut MM cells, providing a rationale to treat RAS mutations in MM. Furthermore, GCK inhibitors might represent an alternative therapy to overcome immunomodulatory drug resistance in MM.
Introduction
Multiple myeloma (MM) is a fatal plasma cell disorder, associated with the accumulation of monoclonal terminally differentiated plasma cells within the bone marrow (BM), renal failure, and bone destruction.1 During the past 16 years, the US Food and Drug Administration approved 12 new therapies with different mechanisms of action for the treatment of MM.2 Despite all this progress, including the introduction of monoclonal antibody therapy, patients still have an overall median survival rate of 5 to 6 years.3,4 Therefore, identification of novel critical pathways and development of specific inhibitors to induce myeloma cell death and/or sensitize myeloma cells to existing treatments are of the highest scientific and clinical merit. RAS mutations play a critical role in many cancers.5 Mutations of the RAS/mitogen-activated protein kinase (MAPK) pathway, including NRAS, KRAS, or BRAF, are found in up to 50% of newly diagnosed MM patients.6,7 The majority of the NRAS, KRAS, and BRAF mutations occur in hotspots causing constitutive activation of the corresponding proteins.7 Although targeting RAS mutations in MM might deepen the response rate and subsequently increase minimal residual disease negativity with the chance of inducing long-term remission, RAS mutations in MM are considered as undruggable.
Germinal center kinase (GCK), also called MAPK kinase kinase kinase 2 (MAP4K2), has emerged as a crucial potential player in the regulation of stress-activated MAPK core signaling pathways,8,9 MAP4Ks belong to the mammalian Ste20-like family of serine/threonine kinases10-12 and play diverse roles in immune cell signaling, immune responses, and inflammation.13 MAP4Ks including MAP4K1/HPK1, MAP4K2/GCK, MAP4K3/GLK, MAP4K4/HGK, MAP4K5/KHS, and MAP4K6/MINK have been reported to induce JNK activation through activating the MAP3K-MAP2K cascade.8,10 MAP4K2/GCK activates JNK by directly binding to and inducing the activation of MEKK1, a MAP3K. GCK also activates another MAP3K, MLK3, which is a specific and potent upstream activator of MKK4 and MKK7. Compared with MEKK1 activation, MLK3 activation relies on GCK-induced phosphorylation, which in turn induces JNK activation.10
GCK is predominantly and highly expressed in the germinal center of B cells,9 which are the regions of the B follicular tissue wherein B lymphocyte differentiation and selection, including immunoglobulin class switch, occur.8 It is known that GCK participates in B-cell differentiation into plasma cells.14 GCK is also expressed in macrophages, which is further increased upon LPS stimulation.15 GCK kinase activity is strongly induced by the stimulation with tumor necrosis factor-α, interleukin-1 (IL-1), or CD40 ligand,16 suggesting that GCK plays essential roles in innate immune response and cell signaling. GCK has also been implicated in cellular processes ranging from stress responses to cell growth, proliferation, and death.8 However, there is only limited knowledge about its role in cancers,13,17-19 and its role in MM remains unknown.
Our studies, for the first time, show that GCK kinase activity is critical for RAS mutated (RASMut) MM cells and regulates MM survival and growth. Because 50% of newly diagnosed patients carry a RAS mutation with a rising percentage in relapsed situation, targeting GCK provides a new approach in the treatment of RASMut multiple myeloma. Furthermore, we showed that GCK inhibition overcomes resistance to IMiD in MM cells, indicating that GCK inhibition holds high therapeutic significance.
Methods
Cell culture and cell selection
MM cell lines RPMI-8226, MM.1S, H929, and U266 were purchased from ATCC. LP-1, KMS12-PE, MOLP-8, SKMM2 and JJN3 were purchased from DSMZ. JIM3 was purchased from Sigma-Aldrich. OPM2 was provided by Dr. Klaus Podar (Karl Landsteiner University of Health Sciences, Krems an der Donau, Austria). The MM cell lines RPMI-8226, MM.1S, H929, U266, and LP-1 were cultured as described previously.20 KMS12-PE, MOLP8, SKMM2, JJN3, and JIM3 were cultured following the manufacturer's protocol. TL4-12 is a selective MAP4K2 inhibitor (Bio-Techne Corporation), named C17 in the original publication.21 The compound was dissolved in dimethyl sulfoxide at a final concentration of 10 mM. Primary human MM cells and human stromal cells were isolated from patient BM samples by Ficoll (GE Healthcare) followed by magnetic separation using CD138+ antibody-specific microbeads according to the manufacturer's protocol (Miltenyi Biotech).22,23 Human BM stromal cells (BMSC) were cultured as previously reported.24 The purity of the BMSCs was assessed by hCD29+/hCD90+/hCD45− staining. All primary samples were obtained after informed consent was given. All studies were approved by the institutional review board of Columbia University Medical Center, New York, NY.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis and western blot analysis
Briefly, the protein was extracted from cells using RIPA buffer containing Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific) and subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and western blotting as described previously.25 The following antibodies were used for immunoblotting: anti-IKZF1 and anti-IKZF3 (Abcam), and anti-CRBN (Sigma-Aldrich), anti-GCK, anti-c-MYC, anti-BCL-6, anti-IRF-4, anti-p-MKK4, anti-p-MKK7, anti-p-JNK, and anti-P53 (Cell Signaling Technology). Anti-β-actin (Sigma-Aldrich) was used to normalize the protein quantity.
Quantitative real-time RT-PCR analysis
For the determination of messenger RNA (mRNA) levels of GCK, total RNAs were isolated from cells using Trizol reagent (Invitrogen, Carlsbad, CA) following the manufacturer’s instructions. Total RNA was converted into complementary DNA using Superscript III RT (Invitrogen). Quantitative reverse transcriptase polymerase chain reaction (RT-PCR) was performed as described before.20 Human GCK, IKZF1, and c-MYC mRNA were analyzed by Taqman real-time RT-PCR (Thermo Fisher Scientific) with β-actin mRNA as the internal control.
Immunohistochemistry analysis of GCK
Formalin-fixed, paraffin-embedded tissue samples of healthy and MM human tissue were obtained from the US Biomax, Inc. Antigen retrieval was achieved using citrate-based retrieval solution, pH 6.1 (Dako). Staining was performed using the EnVision kit (Dako) and cover-slipping using Permount mounting medium (Fisher Scientific).
The following immunohistochemistry antibodies were used: GCK (polyclonal) from Sigma-Aldrich (catalog #HPA007330) and IKZF1 (polyclonal) from Abcam (catalog #26083). The slides were scanned using a high-resolution scanner (Leica SCN400 Slide Scanner) at ×20 magnification. Images were analyzed using Aperio ImageScope software (Aperio Technologies, Inc.). Statistical analysis was performed using the Aperio Positive Pixel Count algorithm in the ImageScope viewing software. An entire tissue section was selected; necrotic regions were excluded with negative gate. The positive pixel count algorithm was used to measure the intensity of each marker (brown signal). Weak and strong positive staining was recorded for the whole tumor section, and the percentage of weak and strong positive pixels was calculated relative to the total number of pixels in the section. P values were calculated using Student t test in GraphPad Prism, version 5, software.
Lentivirus infection, GCK knockdown, and shRNA resistant GCK allele knock-in
To silence GCK expression, human MM cells were transduced with pLKO-Tet-On empty vector lentiviral control (Addgene) or pLKO-Tet-On GCK-targeting short hairpin RNA (shRNA) lentiviral construct (5′-CAGTTTCACCAGGTGAAATTT-3′). Specifically, cells were incubated with lentiviral particles and polybrene (8 μg/mL for 16 hours) and then washed. After 3 days, cells were selected by culturing in puromycin (3 µg/mL) for 7 days. After treatment with doxycycline (400 ng/mL) for 72 hours to activate shRNA, GCK protein and mRNA levels were determined in control and silenced cells by western blotting and real time-PCR, respectively.
To produce GCK-shRNA-resistant GCK sequences, human GCK was cloned from RPMI-8226 cell complementary DNA and inserted into pCDNA6-myc-his6A (Thermo Fisher Scientific, Grand Island, NY) in frame with C-terminal c-myc tag and His6 tag, then subcloned into pCDH-CMV-EF1-eGFP lentiviral vector (System Biosciences). Finally, the GCK shRNA-resistant allele (M5) was generated by Q5 site-directed mutagenesis (New England Biolabs) using the primer pair: forward (5′-AGTCAAATTTGGCGCCCCACGCAGGAAG-3′); reverse (5′-tgatggaactgGATCTCCGAGGGGGTCCTCTC-3′) with pCDH-CMV-GCK(WT)-EF1-eGFP as a template.
GCK kinase dead mutation construct (K to A) with shRNA resistant allele K45A(M5) was generated by site-directed mutagenesis PCR using the primer pair: forward (5′-GGCCGCCGTGGCCATAGTCAAGC-3′); reverse (5′-AGTTCGGACGTGACCGTG-3′) with pCDH-CMV-GCK(M5)-EF1-eGFP as a template. All of the constructs were validated by Sanger sequencing. To knock in GCK, GCK(M5) and K45A(M5) into MM cells expressing inducible shRNA-GCK, MM cells discussed previously were further transduced with the pCDH-EF1-eGFP empty vector, GCK, GCK(M5), or K45A(M5) lentiviral constructs as described previously. Cells were selected by flow cytometry sorting based on the GFP expression. GCK protein expression was determined by western blotting.
Cell proliferation assays
Briefly, MM cells were incubated in 96-well plates for the indicated time. A total of 20 μL of CellTiter 96 Aqueous One Solution Cell Proliferation Assay (MTS; 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt) (Promega) was added to react for 3 hours. Measurement of absorbance of the samples at 490 nm against the background control was performed using the Synergy HT Multi-Detection Microplate Reader (Biotek Instruments, Inc.).
Cell-cycle assays
MM cells (1 × 106 cells/mL) were cultured at 37°C for the indicated time. Cells were then harvested, washed with ice-cold phosphate buffered saline, fixed with 70% ethanol for 1 hour at 4°C, pretreated with RNase (Worthington, Lakewood, NJ) for 30 minutes at 37°C, and then stained with propidium iodine (PI; 20 µg/mL) (Sigma Aldrich). Flow cytometry analysis was conducted on FACS Fortessa cytometer (BD Biosciences) and data were analyzed using FCS Express 6 software (BD Biosciences).
Cell apoptosis assays
The apoptosis of MM cell lines was evaluated by using an Annexin V-FITC Apoptosis Detection Kit I (BD Biosciences, San Jose, CA) per the manufacturer's instructions. MM cells were treated with the appropriate concentration of each agent for the indicated time and incubated with allophycocyanin-conjugated Annexin V and PI for 15 minutes at room temperature in the dark. Flow cytometry analyses were conducted on FACS Fortessa cytometer (BD Biosciences) and data were calculated using FCS Express 6 software (BD Biosciences).
Human MM xenograft mouse model
Female SCID Beige (CB17.Cg-PrkdcscidLystbg-J/Crl) mice were purchased from Charles River Laboratories (Wilmington, MA) at age 6 to 8 weeks with a weight between 20 and 25 g. For human tumor xenograft studies, Tet-on shCNTL or Tet-on shGCK MM.1S-luciferase myeloma cells (5 × 106) in 100 µL phosphate-buffered saline together with an equal volume of Matrigel basement membrane matrix (BD Biosciences) were injected subcutaneously. Sixteen days after implantation, animals were randomized to receive either vehicle (5% sucrose) or doxycycline (1 mg/mL in 5% sucrose) via drinking water for the duration of the study. Mice were weighed twice weekly and observed daily for diarrhea or any changes in behavior and condition. Tumor sizes were measured using a caliper and calculated by the formula: 0.5 × width2 × length, representing the 3-dimensional volume of an ellipse. Tumor sizes were measured twice weekly.24 Tumor growth was monitored by bioluminescence imaging using the IVIS 200 imaging system (Xenogen Corporation). During image acquisition, mice were maintained under general anesthesia with isoflurane.
Lentiviral shRNA silencing of Cereblon
To silence Cereblon (CRBN) expression, MM cells were transduced using pGreenpuro (System Biosciences) lentiviral particles containing CRBN-targeting shRNAs expressing GFP as a selection marker. Stable shRNA transfectants were obtained via cell sorting based on GFP expression. The following target sequences were used: CRBN shRNA (5′-GGAAAGGGAAGCACAGTTT-3′).26 CRBN protein was determined in control and silenced cells by western blotting.
Statistical analyses
Each experiment was repeated at least 3 times, and all quantitative data were presented as mean ± standard deviation. Statistical differences were determined by Student t test. The results were considered statistically significant if P < .05. For the statistical analysis of the differences of the intensity if the immunohistochemistry, Aperio Positive Pixel Count algorithm in the ImageScope viewing software was used.
Results
GCK expression is elevated in MM
Current data are limited to the role of GCK in nonmalignant plasma cell differentiation,12 whereas its expression and function in MM remains unknown. GCK transcription profile of different human tissues provided by genotype-tissue expression portal showed that GCK is expressed at a higher level in Epstein-Barr virus-transformed lymphocytes, spleen cells, and whole blood (Figure 1A). The protein expression of GCK in subtype blood cells provided by the Human Integrated Protein Expression Database showed high expression in monocytes, B-lymphocytes, CD4 T cells, CD8 T cells, natural killer cells, and platelets (Figure 1B). Compared with peripheral blood mononucleated cells (PBMCs) and BMSCs, GCK mRNA levels were significantly higher in primary MM cells and MM cell lines (Figure 1C). Analysis of GCK protein levels by western blotting revealed higher expression in RASMut MM cell lines (MM.1S, RPMI-8226, JJN3, H929, JIM3, and MOLP-8) compared with RASWT cell lines (U266, OPM2, LP-1, KMS12-PE, and SKMM2) (Figure 1D). The high expression of GCK in primary MM was also confirmed by immunohistochemistry staining of patient BM biopsies (Figure 1E, left). By quantitatively comparing the scores of GCK staining of primary MM patient BM (n = 26) and normal donor BM (n = 26), we found the expression of GCK was significantly higher (P < .001) in MM patient BM samples (98%) compared with healthy donor BM samples (47%) (Figure 1E, right).

GCK expression is elevated in MM. (A) The GCK gene and transcript expression values were obtained from the publically available genotype-tissue expression portal on 30 June 2019 and shown as reads per kilobase transcript per million reads (RPKM). (B) The GCK protein expression values were obtained from the Human Integrated Protein Expression Database (HIPED) on 30 June 2019. Parts per million (PPM): each protein entity is enumerated relative to all other protein molecules in the sample. (C) mRNA from a panel of MM cell lines (MM.1S, H929, RPMI-8226, U266), primary MM cell (MM 1 and MM 2), PBMC#1 and PBMC #2, BMSC #1 and BMSC#2, and HEK-293 cells was extracted and GCK mRNA expression was analyzed by quantitative PCR with β-actin as control. GCK mRNA level was calculated relative to its level in HEK-293. (D) RASMut MM cell lines (MM.1S, RPMI-8226, JJN3, H929, JIM3, and MOLP-8) and RASWT cell lines (U266, OPM2, LP-1, KMS12-PE, and SKMM2) were analyzed for GCK protein levels by western blotting of whole cell extracts using β-actin as a loading control. GCK protein levels were quantified using ImageJ software. (E) Immunohistochemistry of paraffin-embedded bone marrow biopsy sections for GCK expression (Brown staining) of normal donor (n = 26) and myeloma patients (n = 26). The slides were scanned using a high-resolution scanner (Leica SCN400 Slide Scanner) at ×40 magnification. Images were analyzed using Aperio ImageScope software (Aperio Technologies, Inc., Vista, CA). Statistical analysis was performed using the Aperio Positive Pixel Count algorithm in the ImageScope viewing software.
GCK expression is elevated in MM. (A) The GCK gene and transcript expression values were obtained from the publically available genotype-tissue expression portal on 30 June 2019 and shown as reads per kilobase transcript per million reads (RPKM). (B) The GCK protein expression values were obtained from the Human Integrated Protein Expression Database (HIPED) on 30 June 2019. Parts per million (PPM): each protein entity is enumerated relative to all other protein molecules in the sample. (C) mRNA from a panel of MM cell lines (MM.1S, H929, RPMI-8226, U266), primary MM cell (MM 1 and MM 2), PBMC#1 and PBMC #2, BMSC #1 and BMSC#2, and HEK-293 cells was extracted and GCK mRNA expression was analyzed by quantitative PCR with β-actin as control. GCK mRNA level was calculated relative to its level in HEK-293. (D) RASMut MM cell lines (MM.1S, RPMI-8226, JJN3, H929, JIM3, and MOLP-8) and RASWT cell lines (U266, OPM2, LP-1, KMS12-PE, and SKMM2) were analyzed for GCK protein levels by western blotting of whole cell extracts using β-actin as a loading control. GCK protein levels were quantified using ImageJ software. (E) Immunohistochemistry of paraffin-embedded bone marrow biopsy sections for GCK expression (Brown staining) of normal donor (n = 26) and myeloma patients (n = 26). The slides were scanned using a high-resolution scanner (Leica SCN400 Slide Scanner) at ×40 magnification. Images were analyzed using Aperio ImageScope software (Aperio Technologies, Inc., Vista, CA). Statistical analysis was performed using the Aperio Positive Pixel Count algorithm in the ImageScope viewing software.
GCK is critical for proliferation and survival of RASMut MM cells
To determine whether GCK protein is required for MM cell proliferation, we transduced MM cells with a robust GCK inducible knockdown (KD) lentivirus pLKO-Tet-On-shGCK in which GCK shRNA expression is induced upon doxycycline treatment. MM.1S and RPMI-8226 are both G12A K-RAS mutated (K-RASG12A) MM cells expressing high levels of GCK, whereas H929 is G13D N-RAS mutated (N-RASG13D) MM cells. Doxycycline-induced Tet-On-shGCK expression resulted in significantly decreased GCK protein level (Figure 2A) and cell proliferation inhibition (P < .01) of MM.1S by 75%, RPMI-8226 by 72%, and H929 by 64% (Figure 2B). In accordance, knockdown of GCK induced cell-cycle arrest in the G0/G1 phase in MM.1S (56.7% in control vs 62.9% in GCK KD), RPMI-8226 (50.1% in control vs 68.1% in GCK KD), and H929 (33.8% in control vs 45.6% in GCK KD) (Figure 2C; supplemental Figure 1, available on the Blood Web site). Importantly, GCK knockdown resulted in a significant increase (P < .001) of apoptotic cells of MM1.S (12% in control vs 32.3% in GCK KD), RPMI-8226 (7.7% in control vs 28.6% in GCK KD), and H929 (8.8% in control vs 66.1% in GCK KD) (Figure 2D; supplemental Figure 1). In RASWT MM cell lines, such as U266 and LP-1, doxycycline-induced Tet-On-shGCK expression resulted in significantly decreased GCK protein level (Figure 2E). In contrast to the high GCK expressing RASMut MM cells, despite a similar knockdown of GCK in RASWT MM cell lines (Figure 2E), decreased GCK protein content failed to induce cell proliferation inhibition and cell apoptosis (Figure 2F-G).
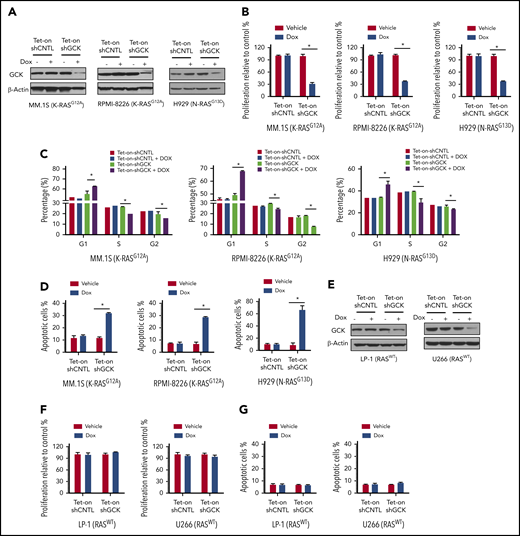
GCK is required for RASMutMM cells survival. (A) MM.1S (K-RASG12A), RPMI-8266 (K-RASG12A), and H929 (N-RASG13D) MM cells were infected by pLKO-Tet-On scramble control (shCNTL) or shGCK lentivirus and selected by puromycin (3 ug/μL) for 1 week. Knockdown of GCK by doxycycline (Dox) treatment (400 ng/mL) for 3 days was confirmed by western blotting. (B-D) Transduced and selected MM.1S, RPMI-8266, and H929 cells were cultured in the presence Dox (400 ng/mL) for 5 days. Cell proliferation was detected by (B) MTS assay, and cells were stained with PI for (C) cell-cycle analysis, or (D) with Annexin V and 7-AAD for apoptosis analysis. (E) U266 and LP-1 RASWT cells were infected by pLKO-Tet-On scramble control (shCNTL) or shGCK lentivirus and selected by puromycin (3 μg/μL) for 1 week. Knockdown of GCK by Dox treatment (400 ng/mL) for 3 days was confirmed by western blotting. (F-G) Transduced and selected U266 and LP-1 RASWT cells were cultured in the presence of Dox (400 ng/mL) to induce shRNA for 5 days. (F) Cell proliferation was detected by MTS assay, and (G) cells were stained with Annexin V and 7-AAD for apoptosis analysis.
GCK is required for RASMutMM cells survival. (A) MM.1S (K-RASG12A), RPMI-8266 (K-RASG12A), and H929 (N-RASG13D) MM cells were infected by pLKO-Tet-On scramble control (shCNTL) or shGCK lentivirus and selected by puromycin (3 ug/μL) for 1 week. Knockdown of GCK by doxycycline (Dox) treatment (400 ng/mL) for 3 days was confirmed by western blotting. (B-D) Transduced and selected MM.1S, RPMI-8266, and H929 cells were cultured in the presence Dox (400 ng/mL) for 5 days. Cell proliferation was detected by (B) MTS assay, and cells were stained with PI for (C) cell-cycle analysis, or (D) with Annexin V and 7-AAD for apoptosis analysis. (E) U266 and LP-1 RASWT cells were infected by pLKO-Tet-On scramble control (shCNTL) or shGCK lentivirus and selected by puromycin (3 μg/μL) for 1 week. Knockdown of GCK by Dox treatment (400 ng/mL) for 3 days was confirmed by western blotting. (F-G) Transduced and selected U266 and LP-1 RASWT cells were cultured in the presence of Dox (400 ng/mL) to induce shRNA for 5 days. (F) Cell proliferation was detected by MTS assay, and (G) cells were stained with Annexin V and 7-AAD for apoptosis analysis.
Knockdown of GCK decreases c-MYC, IKZF1, IKZF3, and BCL6 expression in RASMut MM cells
Knockdown of GCK caused the downregulation of critical transcriptional factors IKZF1/3, c-MYC, and BCL-6 in RASMut MM cells (Figure 3A), all of which are recognized as crucial for MM proliferation,27-30 revealing the novel critical role of GCK. In contrast to the high GCK dependency of RASMut MM cells, knockdown of GCK in U266 RASWT MM cells had no effects on the levels of IKZF1/3, c-MYC and BCL-6 (Figure 3B). The higher sensitivity to GCK KD in RASMut MM cells suggests that targeting GCK is especially effective in MM harboring RAS mutations. To further determine the mechanism of downregulation of IKZF1, we examined the effects of GCK knockdown on the transcription of IKZF1. Real-time PCR analysis showed that GCK knockdown did not downregulate IKZF1 and c-MYC mRNAs (Figure 3C), indicating the absence of GCK-induced regulation of IKZF1/c-MYC at the transcriptional level. It is known that GCK activates JNK1/2 by direct binding and activating MAPK upstream MKK4 and MKK7.11 We analyzed the effects of GCK-knockdown on JNK signaling. Indeed by using inducible Tet-On-shGCK-MM cell line, we found that GCK silencing inhibited the IL-6 induced phosphorylation of MKK4, MKK7, and JNK1/2 in MM.1S cells (Figure 3D). We also investigated whether GCK inhibition abrogates IL-6-induced activation of JNK pathway in RASWT cells. GCK silencing in the RASWT MM cell line LP-1, did not affect IL-6-induced phosphorylation of MKK4, MKK7, and JNK1/2 (Figure 3E). Moreover, coculture with IL-6 partially rescued the cell growth inhibition induced by GCK silencing in MM.1S cells (Figure 3F).
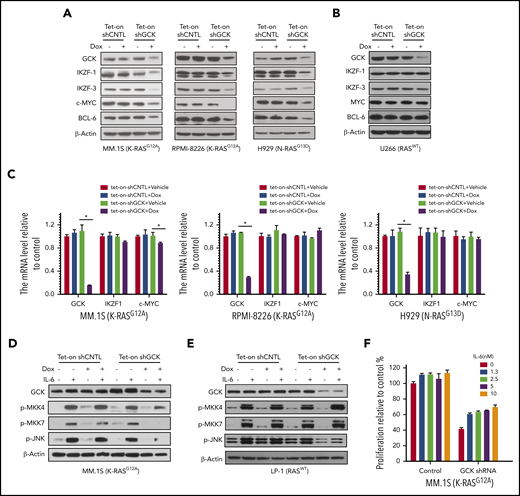
Knock-down of GCK decreases c-MYC, IKZF1, IKZF3, and BCL6 expression in RASMutMM cells. (A-C) MM.1S (K-RASG12D), RPMI-8266 (K-RASG12A), H929 (N-RASG13D), and U266 (RASWT) MM cells were infected by pPLKO-Tet-On scramble control (Tet-On-shCNTL) or sh-GCK lentivirus (Tet-On-shGCK), and selected by puromycin (3 μg/μL) for 1 week. To induce the shRNA, selected cells were treated with Dox 400 ng/mL for 3 days, and analyzed for GCK, IKZF1, IKZF3, c-MYC, and BCL-6 expression by western blotting. (A,B) β-actin was detected as loading control. For mRNA expression of GCK, IKZF1, and c-MYC by quantitative real-time PCR. Data were analyzed according to the ΔΔCt method. Results are shown as mRNA expression relative to control. (C) mRNA levels were normalized with β-actin mRNA expression as control. (D) Tet-On-shCNTL or Tet-On-shGCK MM.1S or (E) LP-1 cells were treated with Dox 400 ng/mL for 36 hours, starved in RPMI-1640 FBS free medium for 12 hours, and treated with IL-6 for 15 minutes. GCK, p-MKK4, p-MKK7, and p-JNK was detected by western blot and β-actin was detected as loading control. (F) Transduced and selected MM.1S (Tet-on-shGCK) cells were cultured with Dox (400 ng/mL) for 2 days, then treated with IL-6 and Dox for 3 days. Cell proliferation was detected by MTS assay.
Knock-down of GCK decreases c-MYC, IKZF1, IKZF3, and BCL6 expression in RASMutMM cells. (A-C) MM.1S (K-RASG12D), RPMI-8266 (K-RASG12A), H929 (N-RASG13D), and U266 (RASWT) MM cells were infected by pPLKO-Tet-On scramble control (Tet-On-shCNTL) or sh-GCK lentivirus (Tet-On-shGCK), and selected by puromycin (3 μg/μL) for 1 week. To induce the shRNA, selected cells were treated with Dox 400 ng/mL for 3 days, and analyzed for GCK, IKZF1, IKZF3, c-MYC, and BCL-6 expression by western blotting. (A,B) β-actin was detected as loading control. For mRNA expression of GCK, IKZF1, and c-MYC by quantitative real-time PCR. Data were analyzed according to the ΔΔCt method. Results are shown as mRNA expression relative to control. (C) mRNA levels were normalized with β-actin mRNA expression as control. (D) Tet-On-shCNTL or Tet-On-shGCK MM.1S or (E) LP-1 cells were treated with Dox 400 ng/mL for 36 hours, starved in RPMI-1640 FBS free medium for 12 hours, and treated with IL-6 for 15 minutes. GCK, p-MKK4, p-MKK7, and p-JNK was detected by western blot and β-actin was detected as loading control. (F) Transduced and selected MM.1S (Tet-on-shGCK) cells were cultured with Dox (400 ng/mL) for 2 days, then treated with IL-6 and Dox for 3 days. Cell proliferation was detected by MTS assay.
Rescue experiments exclude possible off-target effects of GCK shRNA
To exclude possible off-target effects associated with GCK KD, we established an shGCK-resistant GCK allele construct (GCK M5) that rescues the inhibitory effects of shGCK on MM cells (Figure 4A). GCK wild-type (WT) or shGCK-resistant allele M5 lentiviral expression constructs were generated. Tet-On-shGCK-MM.1S cells were transduced with either (1) empty vector (EV), (2) GCK WT, or (3) GCK M5. We hypothesized that the expression level of the shGCK-resistant GCKM5 will not be decreased by shGCK. As expected, doxycycline-induced Tet-On-shGCK expression significantly decreased expression of endogenous GCK and exogenous GCK WT. In contrast, expression of GCK M5 was resistant to shGCK KD (Figure 4B). Consistent with our prior data that GCK regulates IKZF1 and c-Myc expression, shGCK decreased IKZF1 and c-MYC levels in both EV and GCK WT expressing MM cells but failed to do so in the GCK-M5-expressing cells (Figure 4B). In functional assays, GCK KD-induced cell proliferation inhibition was rescued by the introduction of shGCK-resistant GCK-M5 allele (Figure 4C). Similarly, GCK KD induced MM cell apoptosis (Figure 4D) and G1 growth arrest (Figure 4E) in EV- or WT-GCK overexpressing MM cells but failed to induce apoptosis and G1 arrest in the shGCK-resistant GCK-M5 expressing MM cells. These data confirmed that cells expressing shGCK-resistant GCK-M5 were rescued from the shGCK-dependent effects on IKZF1 and c-Myc regulation, MM cell proliferation, G1 growth arrest, and cell apoptosis. These results make possible off-target effects of GCK shRNA highly unlikely. These data further confirmed the critical role of GCK for MM inhibition.

Rescue experiment excluded the possible off-target for GCK shRNA. (A) C-terminal Myc-tagged GCK WT or GCK shRNA-resistant allele M5 expression construct was generated based on PCDH-MCS-EF1-eGFP lentiviral vector as stated in the Methods section. (B) Tet-On-shGCK MM.1S cells were infected with PCDH-EV, PCDH-GCK-myc, or PCDH-GCK(M5)-myc lentiviral particles. Cells were selected by GFP and treated with Dox 400 ng/mL for 2 days to turn on shRNA. Cell lysates were analyzed by western blot for GCK, IKZF1, and c-MYC expression. Selected cells from panel B were cultured with Dox 400 ng/mL for 5 days and cell proliferation was measured by (C) MTS assay, (D) Annexin V and 7-AAD staining for apoptosis analysis, and (E) PI staining for cell-cycle analysis.
Rescue experiment excluded the possible off-target for GCK shRNA. (A) C-terminal Myc-tagged GCK WT or GCK shRNA-resistant allele M5 expression construct was generated based on PCDH-MCS-EF1-eGFP lentiviral vector as stated in the Methods section. (B) Tet-On-shGCK MM.1S cells were infected with PCDH-EV, PCDH-GCK-myc, or PCDH-GCK(M5)-myc lentiviral particles. Cells were selected by GFP and treated with Dox 400 ng/mL for 2 days to turn on shRNA. Cell lysates were analyzed by western blot for GCK, IKZF1, and c-MYC expression. Selected cells from panel B were cultured with Dox 400 ng/mL for 5 days and cell proliferation was measured by (C) MTS assay, (D) Annexin V and 7-AAD staining for apoptosis analysis, and (E) PI staining for cell-cycle analysis.
GCK is critical for MM tumor growth in vivo
Next, we examined whether downregulation of GCK inhibits MM tumor growth in vivo by establishing a Tet-On inducible knockdown system in the SCID/bg mice MM xenograft model. We generated subcutaneous MM xenografts in SCID/bg mice using inducible Tet-On-shCNTL and Tet-On-shGCK MM cells. Doxycycline or vehicle treatment (via drinking water) was started after the tumor was established on day 16 to induce shGCK expression and subsequently GCK knockdown. In contrast to vehicle-treated Tet-On-shGCK or doxycycline-treated MM.1S-Tet-On-shCNTL (control) tumors, doxycycline-treated animals bearing MM.1S-Tet-On-shGCK xenografts showed a significant inhibition (P < .001) of tumor volume by 80% (Figure 5A) and tumor weight by 95% (Figure 5B) after 38 days. Immunohistochemistry staining of tumors confirmed the decrease of GCK expression in doxycycline-treated mice bearing MM.1S-Tet-On-shGCK inducible tumors compared with vehicle-treated MM.1S-Tet-On-shGCK or doxycycline-treated MM.1S-Tet-On-shCNTL tumor (Figure 5C, left). In accordance with our western blot findings, we also observed a decreased expression of IKZF1 expression in GCK knockdown tumors (Figure 5C, right). Next, we expanded the in vivo studies of GCK knockdown on MM tumor progression using bioluminescence (BLI) imaging. For this purpose, MM.1S cells were transduced with firefly luciferase and an inducible GCK knockdown system. Luciferase-expressing GCK inducible knockdown MM.1S cells were subcutaneously injected into SCID/bg mice and the in vivo tumor progression was monitored by BLI. Doxycycline or vehicle treatment was started after the tumor was established on day 16 to induce shGCK and subsequently KD GCK. After 3 weeks treatment of doxycycline, in contrast to MM.1S-Tet-On-shCNTL tumors, animals bearing MM.1S-Tet-On-shGCK xenografts showed a significant inhibition (P < .001) of tumor growth based on the BLI reading by 81% (Figure 5D). These data showing an almost complete abrogation of tumor growth in GCK KD tumors further confirms the critical role of GCK in MM tumorigenesis.
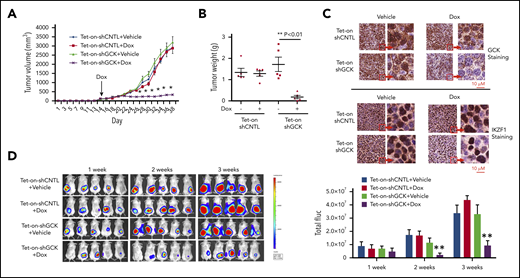
Inhibition of GCK in MM cells abrogates tumor growth in vivo. Tet-on-shCNTL-MM.1S or Tet-On-shGCK-MM.1S cells were injected subcutaneously into SCID/bg mice. Sixteen days after implantation, all mice developed a subcutaneous tumor and were randomized to receive either vehicle (5% sucrose) or Dox (1 mg/mL in 5% sucrose) via drinking water for the duration of study. (A) Subcutaneous tumor growth was measured by using calipers and calculated with the volume formula: 0.5 × long diameter × short diameter.2 Each bar represents the mean ± standard error of the mean (SEM; n = 5). **Indicates significance with P < .01. (B) Mice were euthanized after 38 days. Tumors were excised and weighed. Tumor weights are reported as mean ± SEM (n = 5). **Indicates significance with P < .01. (C) Tumors harvested at the end of the study were fixed in 10% formalin and subsequently processed for immunohistochemical staining for GCK and IKZF1. The slides were scanned using a high-resolution scanner (Leica SCN400 Slide Scanner) at ×40 magnification. (D) SCID/bg mice were injected with Tet-On-shCNTL-MM.1S or Tet-On-shGCK-MM.1S cells expressing luciferase (n = 5). After 1, 2, and 3 weeks, mice received intraperitoneal (3 mg/mouse) d-luciferin 10 minutes before BLI. Bioluminescent signal and grayscale photographic images were acquired using the IVIS Spectrum Bioluminescence and Fluorescence Optical Imaging System and Living Image software.
Inhibition of GCK in MM cells abrogates tumor growth in vivo. Tet-on-shCNTL-MM.1S or Tet-On-shGCK-MM.1S cells were injected subcutaneously into SCID/bg mice. Sixteen days after implantation, all mice developed a subcutaneous tumor and were randomized to receive either vehicle (5% sucrose) or Dox (1 mg/mL in 5% sucrose) via drinking water for the duration of study. (A) Subcutaneous tumor growth was measured by using calipers and calculated with the volume formula: 0.5 × long diameter × short diameter.2 Each bar represents the mean ± standard error of the mean (SEM; n = 5). **Indicates significance with P < .01. (B) Mice were euthanized after 38 days. Tumors were excised and weighed. Tumor weights are reported as mean ± SEM (n = 5). **Indicates significance with P < .01. (C) Tumors harvested at the end of the study were fixed in 10% formalin and subsequently processed for immunohistochemical staining for GCK and IKZF1. The slides were scanned using a high-resolution scanner (Leica SCN400 Slide Scanner) at ×40 magnification. (D) SCID/bg mice were injected with Tet-On-shCNTL-MM.1S or Tet-On-shGCK-MM.1S cells expressing luciferase (n = 5). After 1, 2, and 3 weeks, mice received intraperitoneal (3 mg/mouse) d-luciferin 10 minutes before BLI. Bioluminescent signal and grayscale photographic images were acquired using the IVIS Spectrum Bioluminescence and Fluorescence Optical Imaging System and Living Image software.
GCK kinase activity is required for MM cell growth
Having demonstrated that GCK is required for in vitro MM cell proliferation and in vivo tumor growth, we next addressed whether the GCK kinase activity is required for its function in MM. Lysine 45 is critical for GCK kinase activity (Figure 6A) and a point mutation of K45 to A completely abolishes its kinase activity.31 We overexpressed in MM cells either (1) shGCK-resistant GCK M5 or (2) the kinase dead GCK M5-K45A, both with a C-terminal Myc tag (Figure 6B). Overexpression of GCK M5 K45A kinase-dead mutant significantly reduced MM cell proliferation (Figure 6C). Further, Tet-On-shGCK MM cells were cotransduced with EV, GCK M5, and GCK M5-K45A and treated without or with doxycycline. Doxycycline-induced Tet-On-shGCK expression resulted in significant decrease of GCK protein in EV-transduced cells. In contrast and as expected, expression of exogenous GCK M5 or GCK M5-K45A with C-terminal Myc tag was not affected by shGCK. shGCK induced c-MYC protein downregulation was rescued by shGCK-resistant GCK M5, but not by the kinase dead mutant M5-K45A (Figure 6D). Consistent with the previous data, GCK KD-induced MM cell growth inhibition was abrogated by GCK M5 overexpression. In contrast, GCK kinase-dead mutant M5-K45A failed to recover the decreased cell proliferation induced by GCK downregulation (Figure 6E). Similarly, GCK KD-induced MM cell apoptosis was rescued by GCK M5 overexpression, whereas GCK kinase-dead mutant M5-K45A failed to do so (Figure 6F). Hence, our findings confirmed that GCK kinase activity is required for its role in RASMut myeloma cell survival.

GCK kinase activity is required for MM cell growth. (A) GCK consists of 820 amino acid residues. The kinase domain (in orange) is located at its N terminus. Lysine 45 is critical for GCK kinase activity. Lysine 45 was replaced by alanine for kinase dead mutation. (B-F) MM.1S cells were transduced with EV (PCDH-EV), shRNA-resistant GCK-M5 (PCDH-GCK(M5)-myc), or shRNA-resistant GCK and GCK kinase dead mutation K45A-M5 (PCDH-GCK-K45A(M5)-myc). GFP sorted cells were cultured in the presence of Dox and analyzed for (B) GCK expression by western blot, (C) proliferation by MTS assay, and (D) for GCK and c-MYC expression by western blot. Selected cells from panel D were cultured in the presence of Dox and analyzed for (E) cell proliferation by MTS assay (F) apoptosis by Annexin V and 7-AAD staining.
GCK kinase activity is required for MM cell growth. (A) GCK consists of 820 amino acid residues. The kinase domain (in orange) is located at its N terminus. Lysine 45 is critical for GCK kinase activity. Lysine 45 was replaced by alanine for kinase dead mutation. (B-F) MM.1S cells were transduced with EV (PCDH-EV), shRNA-resistant GCK-M5 (PCDH-GCK(M5)-myc), or shRNA-resistant GCK and GCK kinase dead mutation K45A-M5 (PCDH-GCK-K45A(M5)-myc). GFP sorted cells were cultured in the presence of Dox and analyzed for (B) GCK expression by western blot, (C) proliferation by MTS assay, and (D) for GCK and c-MYC expression by western blot. Selected cells from panel D were cultured in the presence of Dox and analyzed for (E) cell proliferation by MTS assay (F) apoptosis by Annexin V and 7-AAD staining.
Pharmacological blockage of GCK activity inhibits RASMut MM
Because GCK kinase activity is required for MM cell proliferation and survival, we next tested the effects of GCK kinase inhibitor TL4-12 (50% inhibitory concentration = 37 nM)21 on MM. Given that RASMut MM cells have higher GCK expression level and are more sensitive to GCK KD, we compared mutated RAS MM cells with WT RAS MM cells. TL4-12 dose-dependently inhibited the growth of mutated RAS MM cells, with significantly lower 50% inhibitory concentration (JJN3, 1.62 μM; MM1.S, 3.7 μM; H929, 4.4 μM; RPMI-8226, 5.7 μM; and MOLP-8, 10 μM) compared with WT RAS MM cell line (SKMM2, 32 μM; LP-1, 49 μM; U266, 19 μM; and OPM2 and KMS12-PE, no effects) (Figure 7A). Furthermore, TL4-12 dose-dependently decreased IKZF1, c-MYC, and BCL-6 protein expression and increased p53 level in K-RASG12A MM.1S cells, but not in RASWT LP-1 (Figure 7B). In K-RASG12A MM.1S, TL4-12 dose-dependently increased Annexin-V positive cell from 6% (dimethyl sulfoxide) to 13% (1 μM) and 22% (3 μM) (P < .01), respectively. In contrast, in RASWT LP-1 cells, the effects were less potent with only a modest increase of apoptosis at high dose of 3 μM (Figure 7C).
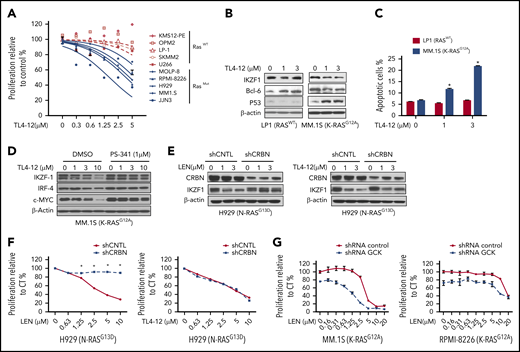
Pharmacological blockage of GCK inhibits MM cell proliferation and induces cell death. (A) RASWT MM cell lines (KMS12-PE, OPM2, LP-1, SKMM-2, and U266) and RASMut MM cell lines (MOLP-8, RPMI-8226, H929, MM.1S, and JJN3) were incubated with TL4-12 for 4 days. Cell proliferation was analyzed by MTS assay. Data represent the mean ± SD of n = 3 experiments. (B) RASWT LP-1 and K-RASG12A MM.1S cells were incubated with TL4-12 for 24 hours to analyze IKZF1, c-MYC, BCL-6, and P53 expression by western blotting. β-actin was detected as loading control. (C) Cells were treated by TL4-12 for 4 days to detect cell apoptosis with Annexin V and 7-AAD staining by flow cytometry assay. (D) MM.1S cells were treated with TL4-12 at the indicated concentrations with or without PS341 (1μM) for 24h. Lysates were analyzed by western blotting to compare the levels of IKZF1, c-MYC, and IRF-4. β-actin expression was probed for loading control. (E-F) N-RASG13D H929 cells were transduced using a lentivirus with control shRNA (shCNTL) or CRBN-shRNA (shCRBN). GFP-sorted cells were cultured in the presence of LEN or TL4-12 for 24 hours to analyze IKZF1, CRBN expression by western blotting (E); and for 4 days for cell proliferation detected by MTS assay (F). (G) Transduced and selected Tet-On-shGCK-MM.1S and Tet-On-shGCK-RPMI-8266 cells were cultured with LEN at the indicated concentrations with or without Dox 400 ng/mL for 3 days and cell proliferation was measured by MTS assay,
Pharmacological blockage of GCK inhibits MM cell proliferation and induces cell death. (A) RASWT MM cell lines (KMS12-PE, OPM2, LP-1, SKMM-2, and U266) and RASMut MM cell lines (MOLP-8, RPMI-8226, H929, MM.1S, and JJN3) were incubated with TL4-12 for 4 days. Cell proliferation was analyzed by MTS assay. Data represent the mean ± SD of n = 3 experiments. (B) RASWT LP-1 and K-RASG12A MM.1S cells were incubated with TL4-12 for 24 hours to analyze IKZF1, c-MYC, BCL-6, and P53 expression by western blotting. β-actin was detected as loading control. (C) Cells were treated by TL4-12 for 4 days to detect cell apoptosis with Annexin V and 7-AAD staining by flow cytometry assay. (D) MM.1S cells were treated with TL4-12 at the indicated concentrations with or without PS341 (1μM) for 24h. Lysates were analyzed by western blotting to compare the levels of IKZF1, c-MYC, and IRF-4. β-actin expression was probed for loading control. (E-F) N-RASG13D H929 cells were transduced using a lentivirus with control shRNA (shCNTL) or CRBN-shRNA (shCRBN). GFP-sorted cells were cultured in the presence of LEN or TL4-12 for 24 hours to analyze IKZF1, CRBN expression by western blotting (E); and for 4 days for cell proliferation detected by MTS assay (F). (G) Transduced and selected Tet-On-shGCK-MM.1S and Tet-On-shGCK-RPMI-8266 cells were cultured with LEN at the indicated concentrations with or without Dox 400 ng/mL for 3 days and cell proliferation was measured by MTS assay,
Immunomodulatory drugs (IMiDs) are part of the backbone of multiple myeloma treatment. IMiDs bind to CRBN, and subsequently induce IKZF1/3 protein degradation that leads to MM cell growth inhibition. Our data showed that GCK knockdown also downregulates IKZF1 level, indicating that IKZF1 is under the regulation of GCK. Concomitant treatment with proteasome inhibitor PS-341 blocked TL4-12-induced IKZF1 downregulation (Figure 7D), suggesting that inhibition of GCK induces IKZF1 protein degradation. However, because IMiD-resistant RPMI-8226 MM cells32 have high expression of GCK and are sensitive to GCK inhibition as shown in Figure 2 and Figure 3A, we hypothesized that the IKZF1 degradation mechanism induced by GCK inhibitor is different from IMiDs and independent of CRBN. To confirm these results, we silenced CRBN in N-RasMut H929 MM cells. CRBN KD was verified by western blot assay and resulted in lenalidomide resistance evidenced by proliferation assay (Figure 7E-F). In contrast, CRBN silencing did not rescue H929 N-RasG13D from TL4-12-induced IKZF1 downregulation and inhibition of proliferation (Figure 7E-F), demonstrating that GCK regulates IKZF1 and cell growth is independent of CRBN. Different from the mechanism of action of IMiD compounds, the inhibition of GCK targets the MAPK cascade and results in IKZF1 degradation independent of CRBN. We further evaluated the combination effects of lenalidomide and GCK silencing in IMiD-sensitive (MM.1S) and resistance (RPMI-8226) MM cell lines. We found that treatment of GCK-silenced MM cells with lenalidomide enhanced the anti-MM effects in both IMiD-sensitive and resistant MM cell lines (Figure 7G).
In summary, our data show that the inhibition of GCK in RASMut MM cells downregulates the phosphorylation and activation of the MAPK cascade, and induces degradation of the critical transcriptional factors including IKZF1, IKZF3, and c-MYC. This results in decreased proliferation and induced apoptosis in RASMut MM cells. Of note, GCK induces IKZF1 degradation via an CRBN independent-mechanism and suggests that targeting GCK may be a very promising strategy to overcome resistance to IMiDs in MM.
Discussion
GCK (MAP4K2) is an essential player in the regulation of stress-activated MAPK core signaling pathways.8 It is predominantly and highly expressed in the germinal center of B cells9 and has been implicated in cellular processes ranging from stress responses to cell growth, proliferation, and death.8,31 More recently, GCK has been reported to play an essential role in pathogen-associated molecular pattern signaling and systemic inflammation through JNK and p38 pathways.13 GCK’s role in cancer as a potential therapeutic target was shown in diffuse large B-cell lymphoma tumors where inhibition of GCK significantly decreased the tumor growth rate in vivo.18 But less is known about the role of GCK in MM.
Our extensive preclinical in vitro and in vivo studies revealed that GCK is critical for MM cell survival and growth and hence represents a promising novel therapeutic target. We demonstrated that doxycycline-induced knockdown of GCK in MM.1S-Tet-On-shGCK xenografts results in an almost complete abrogation of tumor growth in mice. Of the highest importance is our observation that GCK is highly expressed in K- and N-Ras-mutated MM cell lines. Accordingly, K-RasMut (MM.1S, RPMI-8266) and N-RasMut (H929), myeloma cell lines are more sensitive to the GCK inhibition compared with RasWT (LP-1, U266) MM cell lines (Figure 2A, 3A, and 7A). We also analyzed the data of GCK expression in primary MM cells from the MMRF CoMMpass study (IA15). Cases with RASMut (mean = 7.61, n = 257) have significantly decreased GCK expression vs RASWT (mean = 8.36, n = 516) (P = .015) (supplemental Figure 2). Moreover, we found that other MAPK pathway members including BRAF, CRAF, MEKK1, p38, and JNK are all significantly downregulated in the RASMut patients (supplemental Figure 2). We posit that this is due to a feedback mechanism resulting in transcriptional inhibition of the pathway by the overactivated RAS/MAPK signaling. Our data further demonstrated that GCK is targetable by pharmacological inhibition because its cell survival and proliferation regulatory functions are controlled by the GCK kinase activity. Treatment of MM cells with the GCK inhibitor TL4-12 induced cell-cycle arrest and apoptosis in RASMut MM cell lines. Therefore, we concluded that Ras mutations sensitize cells to the therapeutic efficiency of targeting GCK.
GCK induces the phosphorylation and activation of MAPK cascade.33,34 In MM, the MAPK cascade is subject to ubiquitination regulation. MEKK1 displays E3 ubiquitin ligase activity toward ERK1 and ERK2,34 which could triggers subsequently the degradation of the substrate proteins IKZF1, IKZF3, and c-MYC. Indeed, we found that, similar to IMiD compounds, GCK inhibition induces IKZF1, IKZF3, and c-MYC protein degradation in MM cells. Because IKZF1, IKZF3, and activated c-MYC24,25,35 are essential for malignant MM tumor growth, the degradation of these factors significantly inhibited proliferation and induced cell death in both IMiD-sensitive and IMiD-resistant MM cells. More importantly, the knockdown of CRBN in MM led to the resistance to IMiD-induced IKZF1 degradation and MM growth inhibition, but failed to induce resistance to GCK inhibitors. This suggests that the mechanism of GCK inhibitor-induced IKZF1/3 degradation is different from that of IMiDs and is independent of CRBN. Consistently, GCK inhibitions overcome IMiDs resistance in RASMut MM, thereby providing an effective therapeutic approach for relapsed MM patient carrying this mutation. In addition, combination of lenalidomide and GCK silencing enhances the anti-MM effects in both IMiDs sensitive and resistant MM.
Taken together, our findings provide insights into the role of GCK in MM tumorigenesis and the rationale for the clinical evaluation of targeting GCK in patients with RASMut MM. Therefore, the subsequent development of small molecules inhibiting this pathway will address an unmet need for RasMut MM and potentially for other B-cell malignancies, and might affect the management of relapsed and refractory myeloma patients.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Contact the corresponding author for original data.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank the patients and donors for providing samples, and the MMRF CoMMpass study for providing access to patient data.
This research was performed in the CCTI Flow Cytometry Core and supported in part by the Office of the Director of the National Institutes of Health (S10RR027050 and S10OD020056). This work was also supported in part by the National Institutes of Health, National Center for Advancing Translational Sciences (UL1TR001873). S. Li, J.F., and S. Lentzsch were supported by National Institutes of Health, National Cancer Institute (R01CA175313) and Sanofi iAwards. H.M. and M.Y.M. was supported by National Institutes of Health, National Heart, Lung, and Blood Institute (R01HL93716).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Health Science.
Authorship
Contribution: S. Li and J.F conducted experiments; J.Y. contributed to the western blotting assays; H.M. contributed to the cell apoptosis assays; D.B. and M.Y.M. contributed by critically revising the manuscript; and S. Li, J.F., C.M., and S. Lentzsch designed the research studies, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: S. Lentzsch reports Caelum Biosciences equity ownership and membership on Caelum Bioscience's board of directors or advisory committees; consultancy for Janssen, Takeda, GSK, Antengene, Adaptive and Sorrento; and received research funding from Karyopharm and Sanofi. M.Y.M. reports receiving research funding from Ossium Health, Inc and consultancy for Ossium Health. C.M. is a full-time employee of Sanofi. The remaining authors declare no competing financial interests.
Correspondence: Suzanne Lentzsch, Department of Medicine at CUMC, Columbia University, College of Physicians and Surgeons, Herbert Irving Pavilion, R 953, 161 Ft. Washington Ave, New York, NY 10032; e-mail: sl3440@columbia.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal