

BCL-2, discovered as a hallmark of follicular lymphoma in 1985, was the first oncogene described to promote cell survival rather than proliferation. As the founding member of the large BCL-2 family that orchestrates intrinsic apoptosis and as the prime target of venetoclax, it remains a popular therapeutic agent in oncology. Yet, in hematology, as reported in this issue of Blood, its prosurvival sibling MCL-1 has stolen the crown now, as elegantly demonstrated by .1
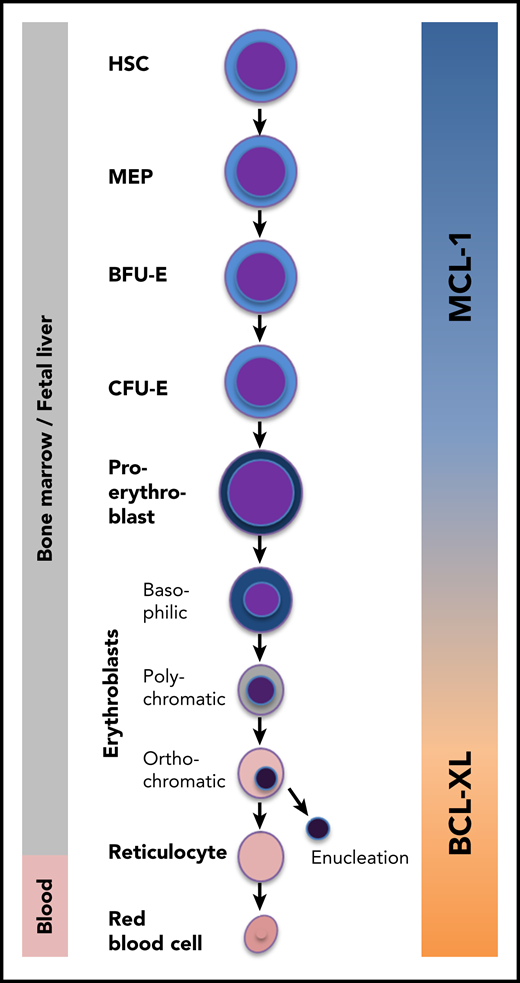
The MCL-1/BCL-XL switch during erythropoiesis. In the bone marrow, hematopoietic stem cells (HSCs) differentiate into red blood cells via multiple successive differentiation stages. Immature stem and progenitor cells and early erythroid progenitors specifically require MCL-1 for survival. During later stages, BCL-XL takes over this function. MEP, megakaryocyte-erythroid progenitor.
The MCL-1/BCL-XL switch during erythropoiesis. In the bone marrow, hematopoietic stem cells (HSCs) differentiate into red blood cells via multiple successive differentiation stages. Immature stem and progenitor cells and early erythroid progenitors specifically require MCL-1 for survival. During later stages, BCL-XL takes over this function. MEP, megakaryocyte-erythroid progenitor.
Turnis et al show that MCL-1 is both sufficient and essential for the production of red blood cells by ensuring the survival of the first hematopoietic cells committed to the erythroid lineage (ie, early burst-forming unit-erythroid [BFU-E] and colony-forming unit-erythroid [CFU-E] cells and proerythroblasts; see figure). Using a mouse model in which both MCL-1 alleles were specifically deleted in cells expressing the receptor for erythropoietin (EPO), the authors observed increased apoptosis in these early cell stages and, consequently, a partial block in erythroid maturation. MCL-1 expression in early erythropoiesis turned out to be vital for the organism because biallelic MCL-1 deletion resulted in anemia and embryonic lethality around E13.5. In vitro differentiation experiments using bone marrow cells derived from adult mice phenocopied this effect: acute MCL-1 deletion resulted in excessive apoptosis of proerythroblasts and early basophilic erythroblasts. In line with the major function of MCL-1 (ie, preventing the activation of the proapoptotic proteins BAX and BAK), concomitant deletion of BAX and BAK fully restored definitive erythropoiesis.
This finding comes as a surprise. Cells committed to the erythroid lineage were believed to require another BCL-2 homolog, BCL-XL, for survival. This assumption was based on results obtained from several independent laboratories: (1) mice lacking BCL-XL died prenatally, and their fetal livers contained a high number of apoptotic erythroid progenitors2 ; (2) in chimeric mice generated by injection of Bcl-x−/− embryonic stem cells into wild-type blastocytes, no mature red blood cells were derived from BCL-XL–deficient hematopoietic cells, whereas myeloid and lymphoid cells were not affected3 ; (3) conditional deletion of BCL-XL in erythroid progenitors using transgenic mice that express the Cre recombinase under the control of the mouse mammary tumor virus long terminal repeat (MMTV-LTR-CRE) caused hemolysis and a left-shifted and increased erythropoiesis. Because BCL-XL–deficient erythroblasts were not depleted, the authors concluded that BCL-XL is only required for the survival of reticulocytes and red blood cells4 ; and (4) complementary work pointed to a role of BCL-XL also in immature erythroid cells, especially when enhanced erythrocyte production is required (eg, after bleeding or in high altitude). Under these conditions, the prosurvival function of BCL-XL is antagonized by BIM, a proapoptotic BCL-2 protein of the BH3-only subgroup. High EPO levels as a consequence of low oxygen foster BCL-XL expression while repressing BIM, thereby increasing the viability of those precursors that proliferate most: proerythroblasts and basophilic erythroblasts. Once the pool of erythrocytes is replenished, EPO levels drop, and the BCL-XL/BIM ratio shifts toward BIM, breaching the apoptosis threshold in a fraction of cells.5 Turnis et al found that EPO also induces MCL-1 expression, albeit only transiently. Thus, EPO-dependent MCL-1 upregulation may contribute to a rapid and efficient increase of red blood cell production during high demand by fostering the survival of early committed progenitors.
The broad implication of this work become evident when considering the expanding list of BH3 mimetics in cancer therapy. Although venetoclax has been approved by the US Food and Drug Administration, an array of promising BH3 mimetic compounds specifically antagonizing MCL-1 or BCL-XL is currently under investigation in early clinical trials for the treatment of various cancers (#NCT02979366, #NCT04178902, #NCT01633541 and others). BH3 mimetics are eagerly awaited by oncologists, but side effects should be expected, especially in the vulnerable hematopoietic system. Opferman et al6 demonstrated earlier that various types of hematopoietic stem and progenitor cells depend on MCL-1 for their survival: acute MCL-1 deletion in adult mice using the Mx-Cre system resulted in lethal bone marrow failure. The new findings of Turnis et al suggest that MCL-1 inhibition as anticancer therapy might not only induce pancytopenia but also unresponsiveness to EPO or other erythropoiesis-stimulating agents. Our own recent work extends the data acquired in mouse models to the human system and confirms the validity of the mouse models. In analogy to murine hematopoiesis, human stem and progenitor cells require MCL-1 for survival,7 whereas erythroid cells are addicted to BCL-XL expression before differentiating into proerythroblasts.8 The available data indicate that developing erythroid cells in humans switch slightly earlier from MCL-1- to BCL-XL dependency than mouse erythroid progenitors. However, because of the lack of robust inducible in vitro systems, human erythropoiesis cannot be studied with the high resolution that is provided by murine models. Refined genetic mouse models, such as those used by Turnis et al, remain of undiminished relevance to understand the hematopoietic system and its vulnerabilities.
How does EPO regulate MCL-1 expression? BCL-XL but not MCL-1 is a direct transcriptional target of GATA1, the master transcription factor of erythropoiesis downstream of the EPO receptor. It is tempting to speculate that the MCL-1/BCL-XL switch during erythropoiesis reflects the GATA2/GATA1 switch that drives erythropoiesis. However, no direct link between GATA2 and MCL-1 has been described so far.
Another important question relates to the nature of the upstream signals that induce apoptosis during physiologic erythroid differentiation, counteracted by MCL-1 or BCL-XL. Obvious candidates for directly antagonizing MCL-1 and/or BCL-XL are the BH3-only proteins BIM, PUMA, and NOXA. BIM has been described to mediate apoptosis induced by EPO deprivation.5 NOXA limits the expansion of erythroblasts during stress erythropoiesis,9 and PUMA causes bone marrow failure on MCL-1 deletion.10 However, the signals upstream of PUMA and NOXA in the hematopoietic system are not yet discovered. Finally, it remains elusive whether and how MCL-1 and BCL-XL contribute to diseases presenting with aregenerative anemia. In this context, it will be interesting to study the roles of these proteins for the pathogenesis of Diamond Blackfan anemia, transient erythroblastopenia of childhood, or pure red cell aplasia.
The results from Turnis et al surely serve as a starting point to solve some of the pressing questions in the fields of hematopoiesis and specifically erythropoiesis.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
