

In this issue of Blood, identify biallelic mutations in RHOG, the gene encoding the small GTPase RhoG, as a novel genetic cause of hemophagocytic lymphohistiocytosis (HLH). This study pinpoints RhoG as an important regulator of lymphocyte cytotoxicity and systematically deciphers the underlying mechanisms.1
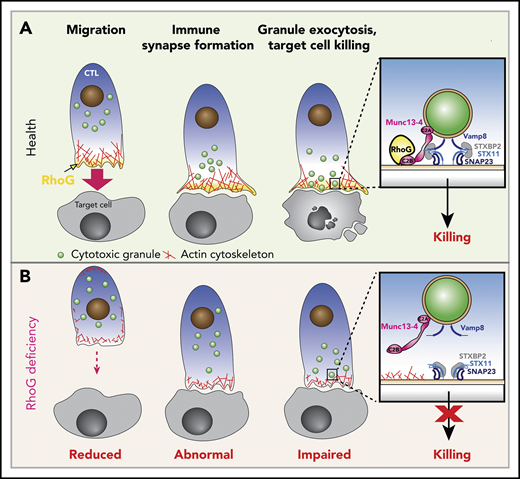
Functions of RhoG in health and effects of RhoG deficiency.(A) Schematic depicting the normal stages of CD8+ T cell recognition and killing of target cells (Health). Upon activation, cytotoxic T lymphocytes (CTL) polarize and migrate toward the target cell via polymerization of actin (shown in red) at the leading edge of the cell, where RhoG (yellow) concentrates. CTLs then make contact with target cells, where they form a stable IS that is driven by the accumulation of actin and RhoG. Meanwhile, CGs (green) are transported to the IS. Granules dock at the IS in an area where actin has cleared through the direct interaction of RhoG with Munc13-4. Inset, The interaction of RhoG with Munc13-4 brings the CGs in contact with the plasma membrane (PM) of the CTL. Subsequent interaction of SNAP receptor (SNARE) proteins present in the CGs (Vamp8) and with those on the PM (Syntaxin 11, SNAP23), along with the SNARE-binding protein STXBP2, enables fusion of the CGs with the PM, releasing granule contents onto the target cell. (B) CTLs lacking expression of RhoG (RhoG deficiency) display reduced polarization and migration, abnormal IS morphology, reduced actin polymerization and clearance zones, and diminished CG exocytosis and target cell killing due to the inability of the granules to properly dock at the IS (inset).
Functions of RhoG in health and effects of RhoG deficiency.(A) Schematic depicting the normal stages of CD8+ T cell recognition and killing of target cells (Health). Upon activation, cytotoxic T lymphocytes (CTL) polarize and migrate toward the target cell via polymerization of actin (shown in red) at the leading edge of the cell, where RhoG (yellow) concentrates. CTLs then make contact with target cells, where they form a stable IS that is driven by the accumulation of actin and RhoG. Meanwhile, CGs (green) are transported to the IS. Granules dock at the IS in an area where actin has cleared through the direct interaction of RhoG with Munc13-4. Inset, The interaction of RhoG with Munc13-4 brings the CGs in contact with the plasma membrane (PM) of the CTL. Subsequent interaction of SNAP receptor (SNARE) proteins present in the CGs (Vamp8) and with those on the PM (Syntaxin 11, SNAP23), along with the SNARE-binding protein STXBP2, enables fusion of the CGs with the PM, releasing granule contents onto the target cell. (B) CTLs lacking expression of RhoG (RhoG deficiency) display reduced polarization and migration, abnormal IS morphology, reduced actin polymerization and clearance zones, and diminished CG exocytosis and target cell killing due to the inability of the granules to properly dock at the IS (inset).
HLH is a rare hyperinflammatory syndrome caused by the dysregulated activation of immune cells that secrete high levels of proinflammatory cytokines and mediate significant tissue damage. The inherited or “primary” forms of HLH are caused by pathogenic germline variants affecting several genes required for the proper function, movement, and release of cytotoxic granules (CGs) by CD8+ T cells and natural killer (NK) cells. Over the last 2 decades, the list of genes responsible for HLH has expanded greatly. Nevertheless, up to 60% of primary HLH cases remain unexplained.2 Furthermore, our understanding of the molecular mechanisms controlling CG movement and release is far from complete.
The work by Kalinichenko et al identifies RHOG as a novel gene in which aberrant function may cause primary HLH and establishes new roles for the RhoG protein in CD8+ T- and NK-cell cytotoxicity. Through exome sequencing, the authors identify compound heterozygous pathogenic variants affecting RHOG in a child with HLH whose lymphocytes exhibited reduced cytotoxic activity and diminished degranulation. Subsequent biochemical and functional analyses of the patient’s lymphocytes and cell lines, modified to either lack RhoG or express activated or dominant-negative forms of RhoG, reveal that the identified RHOG variants abrogate protein expression and impair CD8+ T- and NK-cell killing by negatively affecting cell migration, immune synapse (IS) formation, and CG release (see figure).
RhoG is a small GTPase belonging to the Rho superfamily that regulates key biological processes, including cell proliferation, cytoskeletal dynamics, and vesicle trafficking.3 Although RhoG is known to be important for microtubule and actin cytoskeletal dynamics in various cell types,4 Kalinichenko et al establish a new role for RhoG in modulating the lymphocyte cytoskeleton during cytotoxicity. First, they demonstrate that RhoG deficiency impairs cytoskeletal morphology at the IS, where there is decreased filamentous actin (F-actin) density and reduced formation of actin clearance zones, which are regions of the PM that enable efficient CG exocytosis. The identified defects are associated with decreased activity of Rac1, another small GTPase that functions downstream of RhoG during actin remodeling and cell migration. By treating RhoG-deficient cells with MLN4926, a compound that inhibits Rac1 degradation and thus increases the levels of active Rac1, the authors were able to normalize F-actin levels and cytoskeletal morphology. However, this maneuver only partially rescues degranulation. Together, these results suggest that the activation of Rac1 by RhoG is important for IS formation and function, but they do not fully explain the observed degranulation and cytotoxicity deficiencies observed in the RhoG-deficient cells of the HLH patient under study.
Kalinichenko et al next use interaction proteomics and discover that RhoG physically interacts with Munc13-4, another protein implicated in primary HLH that binds to CGs and facilitates their release.5 Although Munc13-4 has an N-terminal C2A and a C-terminal C2B domain that bind to acidic membrane phospholipids in a calcium-dependent manner,6 how these domains carry out this function has remained elusive. Conversely, RhoG enables the guanine nucleotide exchange factor Trio to bind to membrane phospholipids via the RhoG C-terminal tail.7 Building on these findings, the authors use a series of deletion mutants and demonstrate that it is the interaction of RhoG with Munc13-4 that cooperatively increases the binding affinity of Munc13-4 for membrane phospholipids, where it then presumably enables the tethering and, ultimately, release of CG contents onto the target cell (see figure panel A inset). In RhoG-deficient cells, this process is markedly impaired (see figure panel B inset).
The findings by Kalinichenko et al are notable as they are the first to identify RHOG as a novel genetic cause of primary HLH and they firmly establish RhoG as an essential molecular mediator of lymphocyte cytotoxic function. They also demonstrate the power of “n of 1” studies in which the thorough examination of rare patients with unique phenotypes provides new and important insights into normal physiology and the pathogenesis of human disease. The findings also raise several interesting questions. For example, given the reported findings, should RHOG now be examined in HLH patients who lack mutations in the known genes important for lymphocyte degranulation, such as UNC13D (encoding Munc13-4), STX11, STXBP2, and RAB27A? From a biologic perspective, it is interesting to note that RhoG has critical functions in neurons8 and other cell types,3 yet the RhoG-deficient patient in this report did not display developmental or neurological symptoms. Thus, one must question why the studied patient lacked any broader syndromic manifestations. One possibility is that there are tissue-specific effects of RhoG in immune cells that cannot be compensated by other Rho-family GTPases. Alternatively, and as pointed out by the authors, the rather restrictive phenotype might reflect the tissue-specific expression of critical partner proteins, such as Munc13-4, which is primarily expressed in hematopoietic cells. Although radiographs of the affected patient’s long bones revealed a radiolucent bone lesion, increased sclerosis, and cupping on distal metaphyses, whether these abnormalities are directly related to RhoG deficiency remains to be determined. Future studies of additional patients are needed to understand the full spectrum of clinical manifestations and effects of RhoG deficiency in other cell types, such as neutrophils, mast cells, and platelets, which also use a Munc13-4–dependent exocytosis pathway. Additionally, further studies will be required to elucidate the clinical and biological relevance of other identified germline RHOG variants, as listed in the gnomAD browser (https://gnomad.broadinstitute.org/gene/ENSG00000177105?dataset=gnomad_r2_1). Finally, 2 recent studies describe de novo heterozygous mutations affecting CDC42, the gene encoding Cdc42 (another Rho GTPase), in several patients with neonatal-onset autoinflammation and HLH.9,10 These mutations alter the localization of Cdc42 and impair downstream signaling, leading to defects in cell proliferation, migration, and formation of actin-based structures. Along with the current manuscript, these reports raise the interesting question as to whether Cdc42 and RhoG act through independent pathways to regulate actin cytoskeleton dynamics and CG exocytic machinery, with defects in either pathway leading to distinct yet overlapping hyperinflammatory phenotypes.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
