

In this issue of Blood, describe the mechanism by which monoclonal antibodies to CD44 (anti-CD44) protect against antibody-mediated platelet destruction in murine immune thrombocytopenia (ITP). The antibodies inhibit phagocytosis by targeting the macrophage cell surface by binding to CD44 combined with Fc-dependent blockade of Fcγ-receptors (FcγRs).1
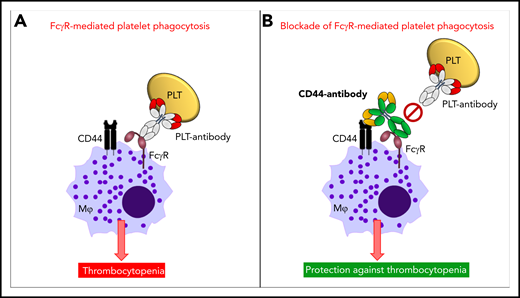
Mechanism of anti-CD44 in protecting against antibody-mediated platelet destruction in ITP. In ITP, antiplatelet autoantibodies opsonize platelets, and these opsonized platelets, through the antibody-Fc region, are recognized by FcγRs on the surface of macrophages. This results in FcγR-mediated platelet phagocytosis by macrophages and in platelet clearance from the circulation resulting in thrombocytopenia (A). Anti-CD44 coengages CD44, as well as FcγRs, on the cell surface of macrophages (in cis), with the anti-CD44 Fc region blocking the macrophage FcγR (Kurlander phenomenon). This prevents FcγR-mediated platelet phagocytosis by macrophages and thus protects against platelet clearance from circulation and development of thrombocytopenia (B). PLT, platelet.
Mechanism of anti-CD44 in protecting against antibody-mediated platelet destruction in ITP. In ITP, antiplatelet autoantibodies opsonize platelets, and these opsonized platelets, through the antibody-Fc region, are recognized by FcγRs on the surface of macrophages. This results in FcγR-mediated platelet phagocytosis by macrophages and in platelet clearance from the circulation resulting in thrombocytopenia (A). Anti-CD44 coengages CD44, as well as FcγRs, on the cell surface of macrophages (in cis), with the anti-CD44 Fc region blocking the macrophage FcγR (Kurlander phenomenon). This prevents FcγR-mediated platelet phagocytosis by macrophages and thus protects against platelet clearance from circulation and development of thrombocytopenia (B). PLT, platelet.
ITP is an autoimmune bleeding disorder characterized by an isolated thrombocytopenia (peripheral blood platelet count <100 × 109/L).2 Clinical symptoms may manifest as petechiae, mucosal bleedings, purpura, and, in rare cases, as intracranial hemorrhages.3 These symptoms negatively impact health-related quality of life. The pathophysiology of ITP is complex, involving multiple factors and cell types.3 In particular, an impairment in CD4+ regulatory T cells and dendritic cells drives the initiation and perpetuation of ITP.3,4 This results in peripheral platelet destruction caused by antiplatelet autoantibody-mediated phagocytosis and/or CD8+ T cell–mediated cytotoxicity.3 Additionally, megakaryocyte function in the bone marrow may be affected by antiplatelet autoantibodies or CD8+ T cells, leading to impaired platelet production.3 Therapeutic strategies in ITP are generally aimed at decreasing platelet-antibody production, blocking clearance of platelets or increasing platelet production by megakaryocytes. Initial treatments for newly diagnosed ITP include corticosteroids, intravenous immunoglobulin (IVIg) and anti-D immunoglobulin.5 Subsequent treatment approaches include rituximab, splenectomy, and thrombopoietin receptor agonists.5 Therapeutic management of patients with ITP remains challenging because of the heterogeneity of disease mechanisms, disease phases (newly diagnosed, persistent, and chronic ITP), and individual patient factors that impact on healthy-related quality of life.
CD44 is a multifunctional cell surface adhesion receptor that is broadly expressed and has been implicated in numerous inflammatory and autoimmune conditions, including in murine antibody-mediated ITP.6 The efficacy of anti-CD44 in preventing ITP has been shown in a murine model of passive antibody-mediated ITP.6 It was subsequently observed that FcγRIIA,6 complement,6 the neonatal Fc receptor (FcRn),7 and the Myd88 signaling pathway8 were all unnecessary for the efficacy of anti-CD44. Thus, the mechanism of action of anti-CD44 in ITP has remained elusive. Norris et al now report that anti-CD44 binds to macrophages and blocks macrophage FcγR-mediated phagocytosis of antibody-opsonized platelets via the so-called Kurlander phenomenon. In a seminal paper in 1983, Roger Kurlander demonstrated that monoclonal antibodies against surface antigens of U-937 cells blocked human immunoglobulin G1 (IgG1) binding dependent of the Fc region of the antibodies, without subsequent FcγR internalization.9 Norris et al elegantly applied a dual approach in their mechanistic dissection of anti-CD44’s mode of action, consisting of an in vitro murine macrophage phagocytosis assay with antibody-opsonized platelets in combination with an in vivo murine model of passive antibody-mediated ITP. They first demonstrated that macrophage pretreatment with anti-CD44 IgG1 or IgG2b inhibited more than 90% of the phagocytosis of platelets opsonized with a monoclonal IgG1 anti-glycoprotein (GP)IIb/IIIa antibody. Blocking macrophage FcγRIII in this phagocytosis assay confirmed the requirement for macrophage FcγRIII in the uptake of IgG1-opsonized platelets. Next, the authors investigated the requirement for the Fc region of anti-CD44 in their phagocytosis assay. For this they made F(ab′)2 and Fc-deglycosylated variants of anti-CD44, for which they confirmed reduced and abrogated binding, respectively, to murine FcγRs. On a functional level, these variants were shown to be incapable of inhibiting antibody-mediated platelet phagocytosis, indicating a critical requirement for the Fc region of anti-CD44 in inhibiting macrophage FcγR-mediated phagocytosis of opsonized platelets. Interestingly, it was further shown that a FcγRIII blocking antibody could no longer bind macrophages that were pretreated with anti-CD44. In contrast, macrophage pretreatment with the F(ab′)2 variant of anti-CD44 did allow the binding of the FcγRIII-blocking antibody to occur, indicating that the Fc region of anti-CD44 is responsible for blocking the macrophage FcγR–IgG binding site. This anti-CD44 blockade did not require interactions with neighboring macrophages, because decreasing macrophage cell density toward isolated macrophages did not impact the blocking effect, supporting a coengagement of anti-CD44 with both macrophage CD44 and FcγRs in cis (see figure). Finally, the authors confirmed that pretreatment with IgG1 anti-CD44 protected mice from ITP development in the passive IgG1-mediated murine model of ITP. In contrast, both the F(ab′)2 and Fc-deglycosylated variants of anti-CD44 were not effective. These findings confirm that the Fc region of anti-CD44 is also required for protection against passive antibody-mediated ITP in vivo. The authors suggest that anti-CD44 may potentially be an attractive alternative for IVIg, which is costly, may induce serious adverse side effects, and often is in short supply.
These findings significantly advance the field while raising new questions and avenues to explore. This works demonstrates the use of anti-CD44 as a preventive strategy for ITP in vitro and in vivo. It is plausible that anti-CD44 may also be able to reverse ongoing platelet phagocytosis and in vivo thrombocytopenia, which will need to be verified. The potential therapeutic applicability of anti-CD44 may be further broadened by researching if monoclonal antibodies directed at other macrophage cell surface antigens than CD44 can also provide prevention of antibody-mediated platelet destruction. Further studies should also explore the likely additional effect of anti-CD44 of blocking the pathogenic effect of C-reactive protein, a ligand for FcγRs, which was previously found to enhance antibody-mediated platelet destruction in phagocytosis assays and in vivo.10 Although the anti-CD44 Fc region appears to be the principle driver of blocking phagocytosis, it may be possible that binding to macrophage cell surface CD44 may induce cellular signaling (eg, potentially affecting activity of Syk), which may make a significant cocontribution in inhibition of phagocytic activity. Finally, translation to the human ITP setting will be important. The first step may be conducting in vitro studies using sera and human cells containing antibodies from patients with ITP.
In sum, Norris et al reveal a novel mechanism of anti-CD44 that targets macrophages through the Kurlander phenomenon, thereby protecting against antibody-mediated platelet clearance in murine ITP. Follow-up work is now highly warranted to further build on these promising results.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal