

Key Points
Abnormal posttranslational palmitoylation of TfR1 results in iron overload in FRDA fibroblasts.
Artesunate, dichloroacetate, and CoA improve TfR1 palmitoylation and reduce iron overload.

Abstract
Friedreich ataxia (FRDA) is a frequent autosomal recessive disease caused by a GAA repeat expansion in the FXN gene encoding frataxin, a mitochondrial protein involved in iron-sulfur cluster (ISC) biogenesis. Resulting frataxin deficiency affects ISC-containing proteins and causes iron to accumulate in the brain and heart of FRDA patients. Here we report on abnormal cellular iron homeostasis in FRDA fibroblasts inducing a massive iron overload in cytosol and mitochondria. We observe membrane transferrin receptor 1 (TfR1) accumulation, increased TfR1 endocytosis, and delayed Tf recycling, ascribing this to impaired TfR1 palmitoylation. Frataxin deficiency is shown to reduce coenzyme A (CoA) availability for TfR1 palmitoylation. Finally, we demonstrate that artesunate, CoA, and dichloroacetate improve TfR1 palmitoylation and decrease iron overload, paving the road for evidence-based therapeutic strategies at the actionable level of TfR1 palmitoylation in FRDA.
Introduction
Friedreich ataxia (FRDA) is a frequent autosomal recessive degenerative disease (1 of 50 000 live births) characterized by progressive spinocerebellar and sensory ataxia, lack of tendon reflexes in the legs, dysarthria, and pyramidal weakness of the inferior limbs, in association with hypertrophic cardiomyopathy and occasionally diabetes mellitus.1 FRDA is primarily caused by GAA repeat expansions within the first intron of FXN, a gene encoding the 210–amino acid mitochondrial protein frataxin. Such expansions account for 98% of FXN-mutant alleles.2 They induce epigenetic silencing, abnormal DNA structures, and aberrant FXN RNA splicing, which together lower the steady-state frataxin level.3 Extensive studies in yeast have shown that YFH1, the yeast homolog of frataxin, is involved in the first step of mitochondrial assembly of iron-sulfur clusters (ISCs), which are subsequently inserted into mitochondrial or cytosolic proteins.4 In YFH1-deleted yeast strains and the hearts of FRDA patients and mice, frataxin deficiency reportedly results in defective ISC-containing proteins, namely cytosolic and mitochondrial aconitases (ACOs), involved in the citric acid cycle and respiratory chain complexes I to III.5,6
Cellular models and pathophysiological studies of FRDA patients suggest a link between frataxin deficiency and altered cellular iron homeostasis. Granular iron deposits have been observed in the hearts7 and brains of FRDA patients,8 as well as in cardiomyocytes of the conditional MCK mouse model reproducing the cardiac phenotype.6 Frataxin deficiency has been shown to increase production of free reactive oxygen species (ROS) in the fibroblasts and plasma of FRDA patients.9 Previous studies have demonstrated that iron accumulates in mitochondria and, to a lesser extent, in the cytosol of Δyfh1 yeast and MCK mice.6,10 However, evidence that frataxin deficiency also causes cytosolic iron dysregulation is inconclusive. Indeed, studies have alternatively shown higher11,12 or lower13-17 ferritin levels in various frataxin deficiency models, and cytosolic and mitochondrial iron content has not been consistently measured.
Here we report on major iron accumulation in both the cytosol and mitochondria of cultured skin fibroblasts from FRDA patients with biallelic FXN GAA expansions. We show that cultured cells were unable to limit iron uptake in our study. Moreover, intracellular coenzyme A (CoA) availability and transferrin receptor 1 (TfR1) palmitoylation were reduced, suggesting that the latter could alter iron uptake and the metal pool in FRDA.
Materials and methods
Cell culture and treatments
Skin fibroblasts from 5 FRDA patients (biallelic FXN GAA repeat expansions) and 3 controls were grown in 5% carbon dioxide at 37°C in Dulbecco’s modified Eagle medium (DMEM; Gibco) supplemented with 4.5 g/L of d-glucose, 110 mg/L of sodium pyruvate, 10% fetal bovine serum (FBS; Gibco), and 200 U/mL of penicillin-streptomycin (Gibco).
Peripheral blood mononuclear cells (PBMCs) isolated from blood samples were grown in serum-free DMEM and 100 µM of ferric ammonium citrate (FAC). Informed consent for diagnostic and research studies was obtained from FRDA patients before their inclusion in the study, in accordance with the Declaration of Helsinki guidelines, and our study was approved by the local institutional review boards (Paris, France).
Protein extraction and immunoblotting
Cells were lysed on ice by scraping with cell lysis buffer (100 nM of tris[hydroxymethyl]aminomethane hydrochloride, pH of 8.3, 600 mM of sodium chloride, 1% Nonidet P-40 Substitute, 20 mM of magnesium chloride, and protease inhibitor cocktail [1×]) and centrifuged at 14 000 rpm for 30 minutes. Western blotting was performed under reducing (2 µM of dithiothreitol [DTT]; denaturation at 95°C for 5 minutes; for frataxin, ferritin, ferroportin [FPN], superoxide dismutase 1 [SOD1], ACO2, ATP5A, PDH-E2, and lipoic acid) or nonreducing (no DTT or heat denaturation; for TfR1, SOD2, and iron regulatory protein 2 [IRP2]) conditions in Laemmli buffer. Antibodies used were: rabbit anti-SOD1 (ab16831; Abcam), rabbit anti-SOD2 (ab13533; Abcam), mouse anti-TfR1 (13-6800; Invitrogen), rabbit anti-ferritin (ab75973; Abcam), rabbit anti-IRP2 (ab181153; Abcam), rabbit anti-frataxin (14147-1-AP; Proteintech), rabbit anti-FPN (NBP1-21502; Novus Biologicals), mouse anti-ACO2 (ab110321; Abcam), mouse anti-ATP5A (ab14748; Abcam), rabbit anti-PDH-E2 (ab172617; Abcam), rabbit anti-lipoic acid (LA; ab58724; Abcam), mouse anti-VDAC1/Porin (ab14734; Abcam), and mouse anti–glyceraldehyde-3-phosphate dehydrogenase antibodies (ab8245; Abcam) and goat anti-rabbit immunoglobulin G (926-68071, LI-COR) or IRDye 680LT goat anti-mouse immunoglobulin G (926-32210, LI-COR).
Detection of mitochondrial ROS
Mitochondrial superoxides were detected by MitoSOX-based flow cytometry (Thermo Fisher Scientific) in accordance with the manufacturer’s protocol.
Subcellular fractionation and isolation of mitochondria
For iron quantification, cytosolic and mitochondrial fractions were obtained using a cell fractionation kit (ab109719; Abcam). For immunoblotting, mitochondria were isolated as previously described.18
Determination of intracellular iron content
Total cellular or subcellular iron content was quantified in culture lysates using a ferrozine-based assay as described.19
RNA extraction and droplet digital polymerase chain reaction
Total RNA extraction and droplet digital polymerase chain reaction was performed as previously described20 using specific fluorescent probes for TFRC (qHsaCIP0033292; Bio-Rad), FTH (dHsaCPE5031151; Bio-Rad), and GUSB complementary DNA detection (qHsaCIP0028142; Bio-Rad).
EMSA
Cytosolic extracts were isolated from fibroblasts with lysis buffer (25 mM of tris[hydroxymethyl]aminomethane hydrochloride, pH of 7.4, 3 mM of magnesium chloride, 40 mM of potassium chloride, 5% glycerol, 0.2% Nonidet P-40, 5 mM of DTT, and Complete EDTA free protease inhibitor cocktail [Roche Applied Science]). After incubation of 20 μg of total protein in 1× electrophoretic mobility shift assay (EMSA) binding buffer supplemented with 5% glycerol, 2 µg of transfer RNA, and 6.25 nM of biotin-labeled iron-responsive element (IRE) for 30 minutes at room temperature (LightShift Chemiluminescent RNA EMSA Kit; Thermo Fisher Scientific), the components were separated with 5% TBE gel (Bio-Rad) at 100 V for 60 minutes and then transferred onto a positively charged nylon membrane (PerkinElmer). After UV cross-linking, biotin signals were detected with horseradish peroxidase–conjugated streptavidin according to the manufacturer’s instructions with the Chemiluminescent Nucleic Acid Detection Module (Thermo Fisher Scientific).
Detection of TfR1 on plasma membrane
Membrane-bound TfR1 was measured by Amnis imaging flow cytometry (Amnis Corporation, MilliporeSigma) as previously described.20
Tf internalization and recycling
Live cell imaging was used to evaluate Tf recycling and internalization. Cells were starved in FBS-free DMEM media for 1 hour. Tf internalization was measured after incubation with Alexa 555–conjugated Tf (Alexa 555–Tf; 12.5 µg/mL) for 5 minutes at 37°C, whereas Tf recycling was evaluated as lost fluorescence after incubation with Tf for 30 minutes at 37°C for at least 20 cells, using spinning-disk microscopy (ZEISS Microscopy) with a 63× oil immersion objective. A 40-minute video of each cell was produced using ZEN software (ZEISS Microscopy). The nuclear region was defined using Icy software and enlarged twice to define the perinuclear region of interest. Masks for this region were then applied on the Alexa 555–Tf channel with the Spot Detector plugin to obtain mean particle fluorescence intensity.
Palmitoylation assay
Palmitoylation assay of TfR1, DMT1, or ZIP14 was performed as previously described20 using anti-TfR1 antibody (136890; Life Technologies), mouse anti-DMT1 (H00004891-M01; Abnova), or rabbit anti-ZIP14 (HPA016508; Sigma-Aldrich) for immunoprecipitation and anti-TfR1 (ab108985; Abcam), anti-DMT1 (20507-1-AP; ProteinTech), or anti-ZIP14 (PA5-21077; Thermo Fisher Scientific) and goat anti-biotin (31852; Thermo Fisher Scientific) for immunoblotting.
CoA assay
CoA was quantified using the Coenzyme-A Assay Kit (ab138889; Abcam) per manufacturer’s instructions.
FXN overexpression
Immortalized fibroblasts were transfected with either GFP or human FXN-GFP complementary DNA cloned in pCCL lentiviral vector as described21 at a final concentration of 1 µg using JetPRIME (Polyplus transfection) in Opti-MEM medium (Gibco). Medium was replaced 6 hours after transfection by fresh DMEM with 10% FBS.
Statistics
Student t tests and 2-way analyses of variance using the Holm-Sidak method were performed with GraphPad Prism (version 6.01) software. Data presented are based on 3 independent experiments (mean ± standard error).
Results
Dysregulation of iron homeostasis in FRDA fibroblasts grown under basal conditions
Our study investigated iron homeostasis in cultures of skin fibroblasts from 5 FRDA patients whose steady-state frataxin levels were significantly lower in comparison with controls (Figure 1A; supplemental Figure 1, available on the Blood Web site). We quantified ferrous and ferric labile iron, as well as the metal complexed with cellular proteins in fibroblasts grown in regular medium (10% FBS), using a ferrozine-based iron assay.19 Iron content was measured in mitochondrial and cytosolic compartments after cellular fractionation. Both mitochondrial and cytosolic iron content was significantly higher in FRDA fibroblasts than in controls (Figure 1B), although the distribution of iron between these compartments was similar across FRDA and control cells (Figure 1C). This contrasts with findings for Δyfh1 yeast strains, where iron accumulated in the mitochondria at the expense of cytosol.10

Iron dysregulation in FRDA fibroblasts. (A) Immunoblot of frataxin in fibroblasts of 5 FRDA patients (P1-P5) and 3 controls (C1-C3) grown in regular medium. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. (B) Iron quantification using the ferrozine-based colorimetric assay in subcellular fractions of control (C* indicates mean of 3 controls) and FRDA (P1-P5) cells grown in regular medium. (C) Relative cytosolic and mitochondrial iron content in controls (C*) and FRDA cells (P1-P5). (D) Immunoblot of several proteins involved in iron homeostasis, ROS defense, and mitochondrial function in control (C1-C3) and FRDA (P1-P5) fibroblasts grown in regular medium. GAPDH was used as a loading control. (E) Quantification of mitochondrial ROS using MitoSOX Red and flow cytometry for control (C*) and FRDA (P1-P5) fibroblasts in regular medium. Data expressed as percentages of MitoSOX+ cells relative to control value. All bar plots show mean ± standard error (n = 3). Student t tests were used to compare patients’ values with control mean. Immunoblotting quantifications are presented in supplemental Figure 1. *P < .05, **P < .01, ***P < .001. ns, nonsignificant.
Iron dysregulation in FRDA fibroblasts. (A) Immunoblot of frataxin in fibroblasts of 5 FRDA patients (P1-P5) and 3 controls (C1-C3) grown in regular medium. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. (B) Iron quantification using the ferrozine-based colorimetric assay in subcellular fractions of control (C* indicates mean of 3 controls) and FRDA (P1-P5) cells grown in regular medium. (C) Relative cytosolic and mitochondrial iron content in controls (C*) and FRDA cells (P1-P5). (D) Immunoblot of several proteins involved in iron homeostasis, ROS defense, and mitochondrial function in control (C1-C3) and FRDA (P1-P5) fibroblasts grown in regular medium. GAPDH was used as a loading control. (E) Quantification of mitochondrial ROS using MitoSOX Red and flow cytometry for control (C*) and FRDA (P1-P5) fibroblasts in regular medium. Data expressed as percentages of MitoSOX+ cells relative to control value. All bar plots show mean ± standard error (n = 3). Student t tests were used to compare patients’ values with control mean. Immunoblotting quantifications are presented in supplemental Figure 1. *P < .05, **P < .01, ***P < .001. ns, nonsignificant.
Homeostasis of cellular iron is regulated by a posttranscriptional mechanism involving IRP1 and IRP2.22 IRP2 is degraded when cytosolic iron concentration increases, causing TfR1 downregulation, which reduces iron uptake, and ferritin upregulation, which promotes iron storage. Western blot analyses of FRDA fibroblasts revealed an expected decrease of IRP2 and increase of ferritin, consistent with cytosolic iron accumulation (Figure 1D; supplemental Figure 1). Paradoxically, dimeric TfR1 steady-state levels actually increased in FRDA fibroblasts, mimicking iron starvation as previously reported upon disruption of the mitochondrial ISC machinery,23 which is not consistent with lower IRP2 levels. Therefore, we hypothesized that TfR1 can escape regulation by IRPs.
Moreover, levels of cytosolic and mitochondrial SOD1 and SOD2 rose (Figure 1D; supplemental Figure 1), suggesting oxidative stress triggered by iron overload. Flow cytometry using MitoSOX consistently revealed a 2.5-fold increase in mitochondrial ROS within FRDA fibroblasts (Figure 1E). Finally, ATP5A, a subunit of oxidative phosphorylation complex V, decreased in FRDA fibroblasts (Figure 1D; supplemental Figure 1), suggesting reduced mitochondrial function despite normal oxidative phosphorylation enzyme activities.5 Altogether these findings point to cellular iron dysregulation associated with cytosolic and mitochondrial iron accumulation in cultured FRDA fibroblasts.
FRDA fibroblasts fail to regulate iron uptake
Cells take up iron primarily as transferrin-bound iron (TBI) and secondarily as non-TBI (NTBI). To better understand the mechanism underlying iron overload, we tested both iron import pathways in FRDA fibroblasts by incubating cells with either increasing concentrations of holo-Tf (1.3-13 mM) or 100 µM of FAC for 72 hours. FAC is a soluble form of Fe3+ iron that enters cells opportunistically via resident transporters or endocytic pathways (NTBI uptake). In both conditions, no FBS was added to deprive the growth medium of TBI. Compared with controls, FRDA fibroblasts not incubated with holo-Tf or FAC exhibited 2.4- to threefold higher iron content (Figure 2A-B). Supplementation with increasing concentrations of holo-Tf gradually increased 1.3- to 1.8-fold the iron content of control fibroblasts, which rapidly reached a plateau, reflecting efficient iron uptake control (Figure 2A). However, increasing concentrations of holo-Tf resulted in rising iron content in FRDA fibroblasts (Figure 2A). After 3 days of incubation with 100 µM of FAC, there was massive iron accumulation in FRDA cells. Indeed, although iron content in control fibroblasts increased 2.3- to threefold in FAC medium, FRDA fibroblasts displayed a 14-fold increase with FAC (Figure 2B). Analysis of the compartmentalization of excess iron revealed higher proportions of mitochondrial iron in FRDA patients (Figure 2C). In FRDA fibroblasts, this iron overload resulted in increased mitochondrial ROS production (Figure 2D), but a similar induction of apoptosis was observed in control and FRDA fibroblasts (supplemental Figure 2A). Taken together, these results suggest that FRDA fibroblasts fail to regulate both TBI and NTBI uptake.
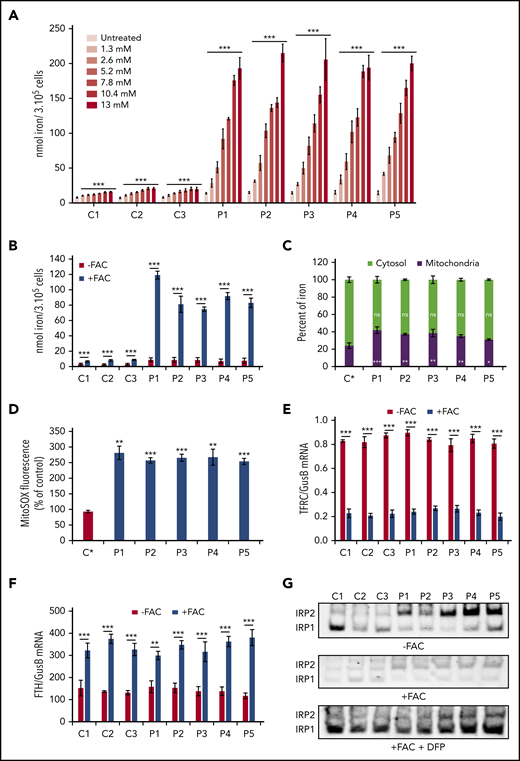
Iron accumulation and posttranscriptional regulation. (A) Iron quantification using the ferrozine-based colorimetric assay in control (C1-C3) and FRDA (P1-P5) fibroblasts grown with increasing concentrations of holo-Tf (1.3-13 mM) for 72 hours. (B) Iron quantification for whole cellular extracts of control (C1-C3) and FRDA (P1-P5) fibroblasts grown in FBS-free DMEM medium under low- (FAC−) or high-iron (100 µM of FAC; FAC+) conditions for 72 hours. (C) Relative cytosolic and mitochondrial iron content in control (C*) and FRDA (P1-P5) fibroblasts grown in FBS-free DMEM with 100 µM of FAC for 72 hours. (D) Quantification of mitochondrial ROS by MitoSOX Red staining in control (C*) and FRDA (P1-P5) fibroblasts grown for 72 hours in high-iron medium (100 µM of FAC). (E-F) Posttranscriptional regulation of iron homeostasis in control (C1-C3) and FRDA (P1-P5) fibroblasts grown in FBS-free DMEM under low- (FAC−) or high-iron (FAC+) conditions. TfR1 (TFRC) (E) and ferritin (FTH) (F) messenger RNAs (mRNAs) were quantified by droplet digital polymerase chain reaction and expressed as ratios to GUSB mRNA. (G) IRP1- and IRP2-IRE binding activity determined by EMSA in low- (FAC−) or high-iron (FAC+) condition with or without deferiprone (DFP; 100 µM) using cytosolic extracts from control (C1-C3) and FRDA (P1-P5) cells. Exposure was 300 seconds for the 3 conditions. Equal amounts of fibroblast protein extracts (20 µg) were assayed. Supplemental Figure 2B-C shows evidence for identification of the 2 bands and immunoblotting quantifications. All bar plots show mean ± standard error (n = 3). Multiple Student t tests (A-D) and 2-way analyses of variance (E-F) were used to compare untreated with treated values. *P < .05, **P < .01, ***P < .001. ns, nonsignificant.
Iron accumulation and posttranscriptional regulation. (A) Iron quantification using the ferrozine-based colorimetric assay in control (C1-C3) and FRDA (P1-P5) fibroblasts grown with increasing concentrations of holo-Tf (1.3-13 mM) for 72 hours. (B) Iron quantification for whole cellular extracts of control (C1-C3) and FRDA (P1-P5) fibroblasts grown in FBS-free DMEM medium under low- (FAC−) or high-iron (100 µM of FAC; FAC+) conditions for 72 hours. (C) Relative cytosolic and mitochondrial iron content in control (C*) and FRDA (P1-P5) fibroblasts grown in FBS-free DMEM with 100 µM of FAC for 72 hours. (D) Quantification of mitochondrial ROS by MitoSOX Red staining in control (C*) and FRDA (P1-P5) fibroblasts grown for 72 hours in high-iron medium (100 µM of FAC). (E-F) Posttranscriptional regulation of iron homeostasis in control (C1-C3) and FRDA (P1-P5) fibroblasts grown in FBS-free DMEM under low- (FAC−) or high-iron (FAC+) conditions. TfR1 (TFRC) (E) and ferritin (FTH) (F) messenger RNAs (mRNAs) were quantified by droplet digital polymerase chain reaction and expressed as ratios to GUSB mRNA. (G) IRP1- and IRP2-IRE binding activity determined by EMSA in low- (FAC−) or high-iron (FAC+) condition with or without deferiprone (DFP; 100 µM) using cytosolic extracts from control (C1-C3) and FRDA (P1-P5) cells. Exposure was 300 seconds for the 3 conditions. Equal amounts of fibroblast protein extracts (20 µg) were assayed. Supplemental Figure 2B-C shows evidence for identification of the 2 bands and immunoblotting quantifications. All bar plots show mean ± standard error (n = 3). Multiple Student t tests (A-D) and 2-way analyses of variance (E-F) were used to compare untreated with treated values. *P < .05, **P < .01, ***P < .001. ns, nonsignificant.
In mammals, IRP signaling governs iron homeostasis by altering TfR1 and ferritin expression through posttranscriptional modification. TFRC mRNA levels were similar in control and FRDA fibroblasts grown in low-iron condition (FAC−) and decreased in high-iron condition (FAC+), suggesting normal downregulation of TFRC transcripts in FRDA (Figure 2E). Ferritin mRNA levels increased in control and FRDA fibroblasts grown under high-iron conditions (Figure 2F), probably as a response to oxidative stress induced by high-iron condition.24 Why similar TFRC mRNA levels were found in control and FRDA fibroblasts is intriguing, because FRDA fibroblasts displayed higher iron content than control cells. We therefore measured the IRP-IRE binding activity in FRDA fibroblasts grown in low- or high-iron media. Decreased IRP1-IRE binding activity was observed in both low- and high-iron conditions compared with controls as expected (Figure 2G; supplemental Figure 2C). Conversely, increased IRP2-IRE binding activity was observed in patients’ cells (Figure 2G; supplemental Figure 2C) as previously reported in ISC-deficient models.14,25-28 This enhanced IRP2-IRE binding activity was expected to increase the amount of TFRC mRNA, which matched control values in FRDA fibroblasts. Finally, control and FRDA fibroblasts grown in high-iron medium with the iron chelator deferiprone (100 µM) displayed similar IRP-IRE binding activity, suggesting that deferiprone reduced iron content to control values in FRDA fibroblasts (Figure 2G; supplemental Figure 2B-C).
At the protein level, iron supplementation in the culture medium (FAC+) increased steady-state ferritin levels in both control and FRDA cells (Figure 3A; supplemental Figure 3) but modestly decreased steady-state IRP2 levels, considering the 14-fold increase of iron content, as demonstrated by western blotting (Figure 3A; supplemental Figure 3). However, steady-state ferritin levels were significantly higher in FRDA fibroblasts (Figure 3A; supplemental Figure 3), consistent with their higher cytosolic iron content and levels of SOD1 and SOD2 (Figure 3A; supplemental Figure 3). Translation of the iron exporter FPN and mitochondrial ACO2 mRNAs is known to be stimulated by iron through decreased IRE-IRP interaction.29 Accordingly, FPN and ACO2 were upregulated in control cells grown in high-iron condition (Figure 3A; supplemental Figure 3). However, FPN and ACO2 were slightly decreased in FRDA fibroblasts (Figure 3A; supplemental Figure 3), a feature ascribed to increased IRP2-IRE binding activity. In contrast, ATP5A was decreased in both control and FRDA fibroblasts grown in high-iron condition (Figure 3A; supplemental Figure 3), suggesting reduced mitochondrial function caused by free radical injury. Under FAC+ conditions, TfR1 levels were lower in controls, preventing iron accumulation, but unexpectedly higher in FRDA cells, despite normal posttranscriptional IRP regulation (Figure 3A; supplemental Figure 3). Increased TfR1 and ferritin levels in the context of iron overload are paradoxical, because stored iron should downregulate TfR1 and metal import. Taken together, these results suggest TfR1 posttranscriptional regulation is normal in FRDA but cooccurs with other dysregulated processes.
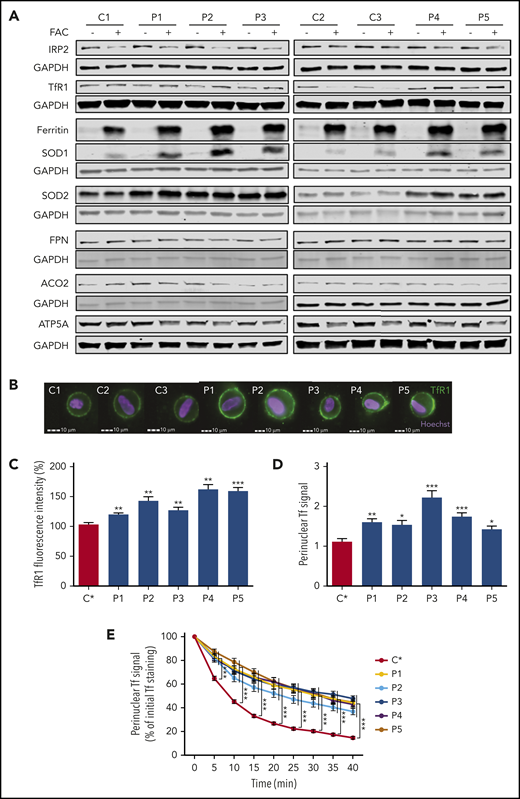
FRDA fibroblasts have impaired iron uptake, accumulate TfR1 at plasma membrane, and exhibit increased transferrin endocytosis. (A) Immunoblots of proteins involved in iron homeostasis, ROS defense, and mitochondrial function in control (C1-C3) and FRDA (P1-P5) cells. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. Immunoblotting quantifications are presented in supplemental Figure 3. (B) Examples of TfR1 labeling in fibroblasts of control (C1-C3) and FRDA (P1-P5) fibroblasts. Cell analysis was based on Hoechst+ signals. Scale bar, 10 µm. (C) Quantification of membrane-bound TfR1 signal in control (C*) and FRDA (P1-P5) fibroblasts grown in regular medium, using IDEAS software (Amnis Corporation). (D) Basal fluorescence intensity of Alexa 555–Tf signal in control (C*) and FRDA (P1-P5) fibroblasts after 5 minutes of Alexa 555–Tf incubation. (E) Relative mean fluorescence intensity of Alexa 555–Tf signal for 40 minutes, after 30 minutes of incubation, for control (C*) and FRDA (P1-P5) fibroblasts. Number of cells acquired was >15 cells per experiment. All bar plots show mean ± standard error (n = 3). Analyses of variance and Student t tests were used to compare patients’ values with control mean. *P < .05, **P < .01, ***P < .001.
FRDA fibroblasts have impaired iron uptake, accumulate TfR1 at plasma membrane, and exhibit increased transferrin endocytosis. (A) Immunoblots of proteins involved in iron homeostasis, ROS defense, and mitochondrial function in control (C1-C3) and FRDA (P1-P5) cells. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. Immunoblotting quantifications are presented in supplemental Figure 3. (B) Examples of TfR1 labeling in fibroblasts of control (C1-C3) and FRDA (P1-P5) fibroblasts. Cell analysis was based on Hoechst+ signals. Scale bar, 10 µm. (C) Quantification of membrane-bound TfR1 signal in control (C*) and FRDA (P1-P5) fibroblasts grown in regular medium, using IDEAS software (Amnis Corporation). (D) Basal fluorescence intensity of Alexa 555–Tf signal in control (C*) and FRDA (P1-P5) fibroblasts after 5 minutes of Alexa 555–Tf incubation. (E) Relative mean fluorescence intensity of Alexa 555–Tf signal for 40 minutes, after 30 minutes of incubation, for control (C*) and FRDA (P1-P5) fibroblasts. Number of cells acquired was >15 cells per experiment. All bar plots show mean ± standard error (n = 3). Analyses of variance and Student t tests were used to compare patients’ values with control mean. *P < .05, **P < .01, ***P < .001.
Increased amount of membrane TfR1 and delayed transferrin recycling in FRDA fibroblasts
We hypothesized that higher steady-state TfR1 levels could be related to an accumulation of homodimeric membrane TfR1 in FRDA cells, as previously observed in neurodegeneration with brain iron accumulation.20 TfR1 was quantified by immunofluorescence using the Amnis ImageStreamX Mark II, which combines flow cytometry with detailed cell imaging and functional analysis. More TfR1 was found on the surface of FRDA fibroblasts than on control cells grown in regular medium (Figure 3B-C). Thus, despite iron overload and normal TFRC transcript amount, patient fibroblasts paradoxically accumulated membrane TfR1, preventing downregulation of iron uptake. Live imaging via spinning-disk microscopy was then used to assess Tf-TfR1 complex endocytosis through evaluation of perinuclear fluorescence intensity. FRDA cells exhibited a significantly higher Tf signal compared with controls (Figure 3D), suggesting greater Tf-TfR1 internalization. Tf-TfR1 recycling was then measured as the loss of cell-associated fluorescence over time. Control fibroblasts displayed rapid loss of Tf staining ascribed to Tf recycling (Figure 3E; supplemental Movie 1). By contrast, Tf recycling was significantly delayed in FRDA fibroblasts, because the specific perinuclear signal failed to weaken after 10 minutes of Alexa 555–Tf incubation and was still delayed later (Figure 3E; supplemental Movie 2). The combination of greater Tf-TfR1 endocytosis and delayed recycling was expected to increase iron release from early endosomes and contribute to iron overload in frataxin-deficient cells.
Artesunate rescues TfR1 palmitoylation defect in FRDA fibroblasts
TfR1 undergoes posttranslational modification by covalent attachment of S-acyl radicals to Cys62 and Cys67 via thioester bonds, with palmitate being the predominant fatty acid donor.30 Decreased palmitoylation has been previously shown to induce accumulation of TfR1 at cell membranes and increase its endocytosis.20,30 Studying FRDA cells through acyl-biotin exchange, we observed a dramatic fall in TfR1-biotin levels, reflecting defective TfR1 palmitoylation (only 16% to 22% of control levels; Figure 4A; supplemental Figure 4A). This result suggests that frataxin deficiency severely alters TfR1 palmitoylation for reasons still unknown.
![Artesunate attenuates cellular iron dysregulation in FRDA fibroblasts. (A) Biotin immunoblots correlating with extent of TfR1 palmitoylation in control (C1-C3) and FRDA (P1-P5) fibroblasts grown in regular medium (artesunate−) or supplemented with 25 µM of artesunate (artesunate+) for 48 hours. For each condition, the topmost panel shows the palmitoylated TfR1 level (immunoprecipitated [IP]: biotin) and the panel below shows the IP amount of TfR1 (IP: TfR1). IP TfR1 was used as a loading control for each condition. Immunoblotting quantifications of palmitoylated and steady-state TfR1 levels are given in supplemental Figure 4A-D. (B) Quantification of membrane-bound TfR1 signal for control (C1-C3) and FRDA (P1-P5) fibroblasts, grown for 48 hours with or without 25 µM of artesunate supplementation, using IDEAS software (Amnis Corporation). Examples of TfR1 labeling in control and FRDA fibroblasts are given in supplemental Figure 3E. (C) Iron quantification using ferrozine-based colorimetric assay in control (C1-C3) and FRDA (P1-P5) fibroblasts grown in regular medium, with or without 25 µM of artesunate for 48 hours. (D) Immunoblot analysis of proteins involved in iron homeostasis and ROS defense in control (C1-C3) and FRDA (P1-P5) fibroblasts grown in regular DMEM medium, with or without 25 µM of artesunate, for 48 hours. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. Immunoblotting quantifications are given in supplemental Figure 4F. All bar plots show mean ± standard error (n = 3). Two-way analyses of variance were used to compare untreated with treated values. *P < .05, **P < .01, ***P < .001. ns, nonsignificant.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/15/10.1182_blood.2020006987/1/m_bloodbld2020006987f4.png?Expires=1769164871&Signature=gconKB1kuWBlpYwCcpANyI8IY6afnZdW1Nkhk63s3-SSwCgBAQKvNP8ucxcpCRt~W4E2Woe80n8~CRLIR1KaMT96VYn50aQtS0j2CIvHHBdVd2XALCi85ezBVVOXoJmzmcjdYPFUwutccsp0lbYg9REVAACZamqDdNHRfeq8fFOiNFgSj15vhYyI4CnsNtQ720g6sCR3t3iPnlvEd5wh~At2QVkx4cNnIGnKzYp7G8BIsmgFD9nelp7MwCj-Dw5M83NOvea~agJFKCcOgTgy5ggIDrYKOx~-6iS~WeupyhZWB2pY3I3won1OQf4Xg1CCY1h2a0XfvN5yuArkDbw4Lw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Artesunate attenuates cellular iron dysregulation in FRDA fibroblasts. (A) Biotin immunoblots correlating with extent of TfR1 palmitoylation in control (C1-C3) and FRDA (P1-P5) fibroblasts grown in regular medium (artesunate−) or supplemented with 25 µM of artesunate (artesunate+) for 48 hours. For each condition, the topmost panel shows the palmitoylated TfR1 level (immunoprecipitated [IP]: biotin) and the panel below shows the IP amount of TfR1 (IP: TfR1). IP TfR1 was used as a loading control for each condition. Immunoblotting quantifications of palmitoylated and steady-state TfR1 levels are given in supplemental Figure 4A-D. (B) Quantification of membrane-bound TfR1 signal for control (C1-C3) and FRDA (P1-P5) fibroblasts, grown for 48 hours with or without 25 µM of artesunate supplementation, using IDEAS software (Amnis Corporation). Examples of TfR1 labeling in control and FRDA fibroblasts are given in supplemental Figure 3E. (C) Iron quantification using ferrozine-based colorimetric assay in control (C1-C3) and FRDA (P1-P5) fibroblasts grown in regular medium, with or without 25 µM of artesunate for 48 hours. (D) Immunoblot analysis of proteins involved in iron homeostasis and ROS defense in control (C1-C3) and FRDA (P1-P5) fibroblasts grown in regular DMEM medium, with or without 25 µM of artesunate, for 48 hours. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. Immunoblotting quantifications are given in supplemental Figure 4F. All bar plots show mean ± standard error (n = 3). Two-way analyses of variance were used to compare untreated with treated values. *P < .05, **P < .01, ***P < .001. ns, nonsignificant.
Artesunate attenuates cellular iron dysregulation in FRDA fibroblasts. (A) Biotin immunoblots correlating with extent of TfR1 palmitoylation in control (C1-C3) and FRDA (P1-P5) fibroblasts grown in regular medium (artesunate−) or supplemented with 25 µM of artesunate (artesunate+) for 48 hours. For each condition, the topmost panel shows the palmitoylated TfR1 level (immunoprecipitated [IP]: biotin) and the panel below shows the IP amount of TfR1 (IP: TfR1). IP TfR1 was used as a loading control for each condition. Immunoblotting quantifications of palmitoylated and steady-state TfR1 levels are given in supplemental Figure 4A-D. (B) Quantification of membrane-bound TfR1 signal for control (C1-C3) and FRDA (P1-P5) fibroblasts, grown for 48 hours with or without 25 µM of artesunate supplementation, using IDEAS software (Amnis Corporation). Examples of TfR1 labeling in control and FRDA fibroblasts are given in supplemental Figure 3E. (C) Iron quantification using ferrozine-based colorimetric assay in control (C1-C3) and FRDA (P1-P5) fibroblasts grown in regular medium, with or without 25 µM of artesunate for 48 hours. (D) Immunoblot analysis of proteins involved in iron homeostasis and ROS defense in control (C1-C3) and FRDA (P1-P5) fibroblasts grown in regular DMEM medium, with or without 25 µM of artesunate, for 48 hours. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. Immunoblotting quantifications are given in supplemental Figure 4F. All bar plots show mean ± standard error (n = 3). Two-way analyses of variance were used to compare untreated with treated values. *P < .05, **P < .01, ***P < .001. ns, nonsignificant.
Artesunate is known to alter cellular iron homeostasis by increasing TfR1 palmitoylation and reducing membrane TfR1.31 Addition of 25 µM of artesunate to the culture medium enhanced TfR1 palmitoylation and significantly decreased the steady-state level of TfR1 in FRDA fibroblasts (Figure 4A; supplemental Figure 4A-D). Furthermore, flow cytometric imaging showed that artesunate lowered membrane TfR1 levels by 15% to 30% in FRDA fibroblasts (Figure 4B; supplemental Figure 4E) but raised them 1.1- to 1.3-fold in control cells (Figure 4B; supplemental Figure 4E). Importantly, artesunate significantly decreased the iron overload in FRDA fibroblasts (Figure 4C).
Finally, immunoblot analyses confirmed that artesunate lowered TfR1 levels in FRDA cells (Figure 4D; supplemental Figure 4F), whereas IRP2 levels increased, in parallel with decreased steady-state ferritin concentrations (Figure 4D; supplemental Figure 4F). This is to be expected with lower cellular iron content. Near return of SOD2 to control levels was consistently observed (Figure 4D; supplemental Figure 4F). In control cells, artesunate slightly increased TfR1 (Figure 4D; supplemental Figure 4F), paralleling the increase in membrane TfR1 (Figure 4B; supplemental Figure 4E). However, it did not alter intracellular iron content (Figure 4C). These findings illustrate the impact of TfR1 palmitoylation on cellular iron content and suggest a possibly beneficial effect of artesunate on cellular iron pools in FRDA.
Impaired TfR1 palmitoylation is related to defective pyruvate dehydrogenase lipoylation in FRDA fibroblasts
Lipoic acid synthase, a key enzyme of LA synthesis, is a (4Fe-4S) cluster–containing protein severely affected by deficient assembly of the ISC machinery.32 LA is a cofactor of several mitochondrial proteins, including dihydrolipoamide acetyltransferase (PDH-E2), 1 of the 3 pyruvate dehydrogenase (PDH) subunits. Biallelic mutations in genes involved in ISC biogenesis (NFU1, IBA57, and ISCA2) alter lipoylation of PDH-E2 and cause a secondary decrease in PDH activity.33 Moreover, FXN depletion in mice and knockdown of FXN in HeLa cells have been shown to greatly disrupt PDH lipoylation.16,34 We likewise observed altered lipoylation of PDH-E2 and a lower steady-state level of PDH-E2 in FRDA fibroblasts (Figure 5A; supplemental Figure 5A).
![FRDA fibroblasts display CoA and PDH lipoylation defects. (A) Immunoblots of PDH-E2 and lipoylated PDH-E2 (PDH-E2-LA) in mitochondria-enriched extracts from control (C1-C3) and FRDA (P1-P5) grown in regular medium for 72 hours, with or without 5 mM of dichloroacetate (DCA). Porin was used as a loading control. Immunoblotting quantifications are given in supplemental Figure 5A. (B) Total CoA content in control (C1-C3) and FRDA (P1-P5) cells grown in untreated regular medium, with 25 µM of CoA or 5 mM of DCA, for 72 hours. (C) Biotin immunoblots correlating with extent of TfR1 palmitoylation in control (C1-C3) and FRDA (P1-P5) fibroblasts grown in untreated regular medium or supplemented with either 25 µM of CoA or 5 mM of DCA for 72 hours. For each condition, the topmost panel shows palmitoylated TfR1 levels (immunoprecipitated [IP]: biotin), the panel below shows the IP amount of TfR1 (IP: TfR1), and IP TfR1 was used as a loading control. Immunoblotting quantifications of palmitoylated and steady-state levels of TfR1 are given in supplemental Figure 5B-C. (D) Immunoblot analysis of control (C1-C3) and FRDA (P1-P5) immortalized fibroblasts overexpressing wild-type FXN complementary DNA (cDNA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. Immunoblotting quantifications are presented in supplemental Figure 6A-C. (E) Iron quantification using ferrozine-based colorimetric assay in control (C1-C3) and FRDA (P1-P5) immortalized fibroblasts overexpressing wild-type FXN cDNA. Plots show mean ± standard error (n = 3). Two-way analyses of variance were used to compare untreated with treated values. *P < .05, ***P < .001. ns, nonsignificant.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/15/10.1182_blood.2020006987/1/m_bloodbld2020006987f5.png?Expires=1769164871&Signature=og3UbKKklkyqzJZkdIoY0aNUgD4HuytJhaWgPNULWh59p83OAPPWnBsLWwszRsuIN2tHiZkqEgc2UOiuqXtujAtgiIeyC9jfVpfuZUo3hCk7NFxtGO4mUZeF1mhZQJYjb~fVLX09CT4Wj0qjsfeHFp5fQvYnIMqbcfLZCOjQaQsOUQjfq-Dg2zyy5PSPblXCX9CHs0sKT1iKD5H0iIMSOnsJ8UERXms1qFuMzsaLwesQZCslGdpx-TTa9cl6XBg4aw3B-YgX6ihQc-wjIO-0KmgzQebzYujOnltXc2-fAZ3otgjXnb2Ove83DLhrfaHrRh1MaaGS~9IPlDWduS7r~A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
FRDA fibroblasts display CoA and PDH lipoylation defects. (A) Immunoblots of PDH-E2 and lipoylated PDH-E2 (PDH-E2-LA) in mitochondria-enriched extracts from control (C1-C3) and FRDA (P1-P5) grown in regular medium for 72 hours, with or without 5 mM of dichloroacetate (DCA). Porin was used as a loading control. Immunoblotting quantifications are given in supplemental Figure 5A. (B) Total CoA content in control (C1-C3) and FRDA (P1-P5) cells grown in untreated regular medium, with 25 µM of CoA or 5 mM of DCA, for 72 hours. (C) Biotin immunoblots correlating with extent of TfR1 palmitoylation in control (C1-C3) and FRDA (P1-P5) fibroblasts grown in untreated regular medium or supplemented with either 25 µM of CoA or 5 mM of DCA for 72 hours. For each condition, the topmost panel shows palmitoylated TfR1 levels (immunoprecipitated [IP]: biotin), the panel below shows the IP amount of TfR1 (IP: TfR1), and IP TfR1 was used as a loading control. Immunoblotting quantifications of palmitoylated and steady-state levels of TfR1 are given in supplemental Figure 5B-C. (D) Immunoblot analysis of control (C1-C3) and FRDA (P1-P5) immortalized fibroblasts overexpressing wild-type FXN complementary DNA (cDNA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. Immunoblotting quantifications are presented in supplemental Figure 6A-C. (E) Iron quantification using ferrozine-based colorimetric assay in control (C1-C3) and FRDA (P1-P5) immortalized fibroblasts overexpressing wild-type FXN cDNA. Plots show mean ± standard error (n = 3). Two-way analyses of variance were used to compare untreated with treated values. *P < .05, ***P < .001. ns, nonsignificant.
FRDA fibroblasts display CoA and PDH lipoylation defects. (A) Immunoblots of PDH-E2 and lipoylated PDH-E2 (PDH-E2-LA) in mitochondria-enriched extracts from control (C1-C3) and FRDA (P1-P5) grown in regular medium for 72 hours, with or without 5 mM of dichloroacetate (DCA). Porin was used as a loading control. Immunoblotting quantifications are given in supplemental Figure 5A. (B) Total CoA content in control (C1-C3) and FRDA (P1-P5) cells grown in untreated regular medium, with 25 µM of CoA or 5 mM of DCA, for 72 hours. (C) Biotin immunoblots correlating with extent of TfR1 palmitoylation in control (C1-C3) and FRDA (P1-P5) fibroblasts grown in untreated regular medium or supplemented with either 25 µM of CoA or 5 mM of DCA for 72 hours. For each condition, the topmost panel shows palmitoylated TfR1 levels (immunoprecipitated [IP]: biotin), the panel below shows the IP amount of TfR1 (IP: TfR1), and IP TfR1 was used as a loading control. Immunoblotting quantifications of palmitoylated and steady-state levels of TfR1 are given in supplemental Figure 5B-C. (D) Immunoblot analysis of control (C1-C3) and FRDA (P1-P5) immortalized fibroblasts overexpressing wild-type FXN complementary DNA (cDNA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. Immunoblotting quantifications are presented in supplemental Figure 6A-C. (E) Iron quantification using ferrozine-based colorimetric assay in control (C1-C3) and FRDA (P1-P5) immortalized fibroblasts overexpressing wild-type FXN cDNA. Plots show mean ± standard error (n = 3). Two-way analyses of variance were used to compare untreated with treated values. *P < .05, ***P < .001. ns, nonsignificant.
Acetyl-CoA, the product of the PDH reaction, is the sole donor of acetyl groups for palmitoyl transferases. We hypothesized that frataxin deficiency could limit the CoA pool and secondarily affect TfR1 palmitoylation. We noted 1.6- to 2.1-fold lower total cellular CoA content in FRDA fibroblasts relative to control cells (Figure 5B), confirming that FXN mutations limited the CoA pool. Considering that mitochondrial acetyl-CoA is the major source of cytosolic acetyl-CoA in normal cells,35 the decrease in total CoA should reflect a reduction of both mitochondrial and cytosolic CoA/acetyl-CoA pools.
Supplementing cultured cells with CoA (25 µM for 72 hours) resulted in increased TfR1 palmitoylation in FRDA fibroblasts (Figure 5C; supplemental Figure 5B). CoA supplementation also decreased steady-state TfR1 levels in patient cells (Figure 5C; supplemental Figure 5C). Furthermore, addition of DCA (5 mM for 72 hours), an inhibitor of PDH kinase known to augment pyruvate oxidation and the acetyl-CoA pool, significantly raised FRDA fibroblast CoA content (Figure 5B) and restored PDH lipoylation (Figure 5A; supplemental Figure 5A). DCA also significantly lowered steady-state levels of TfR1 in FRDA fibroblasts (Figure 5C; supplemental Figure 5C). These results collectively suggest that frataxin deficiency leads to secondary reduction of the CoA pool required for palmitoylation and endocytosis of TfR1.
Finally, we overexpressed wild-type frataxin in control and FRDA immortalized fibroblasts, and colocalization of frataxin and MitoTracker indicated mitochondrial targeting (Figure 5D; supplemental Figure 6A,D). Whereas the steady-state TfR1 level was unchanged in control cells, it significantly decreased to control values in FRDA fibroblasts overexpressing frataxin in mitochondrial compartments and reached control values (Figure 5D-E; supplemental Figure 6A-C). Consistently, iron content returned to control values in FRDA fibroblasts (Figure 5E), establishing a clear relationship between frataxin defect, TfR1 accumulation, and iron overload.
Role of palmitoylation in TBI and NTBI uptake
Finally, we wanted to know if defective palmitoylation of other proteins might also contribute to iron uptake dysregulation in FRDA. Cells were grown in FAC medium supplemented with 2-bromopalmitate (2BP; 100 µM), a well-known inhibitor of protein S-palmitoylation, for 16 hours. Remarkably, 2BP substantially increased iron overload in both FRDA fibroblasts and control cells (Figure 6A), suggesting a role for protein palmitoylation in NTBI uptake. We investigated the palmitoylation of 2 NTBI transporters, DMT1 and ZIP14, by acyl-biotin exchange and observed a significant decrease of their palmitoylation in FRDA fibroblasts (DMT1, 50% and ZIP14, 40% of controls values; Figure 6B; supplemental Figure 7A-B), possibly altering NTBI uptake capacity. DMT1 and ZIP14 steady-state levels were also lowered in FRDA fibroblasts. To investigate the respective parts played by TBI and NTBI uptake in iron overload, we quantified the effects of holo-Tf and FAC on iron content in cells supplemented with CoA, DCA, or artesunate. Reduced iron overload was seen under all conditions in FRDA cells, but not in controls (except for artesunate with FAC; Figure 6C-F). These observations are consistent with the view that iron overload in FRDA is triggered by both TBI and NTBI uptake and limited by expansion of the acetyl-CoA pool.
![Enhancement of TBI and NTBI uptake in FRDA fibroblasts. (A) Iron quantification using the ferrozine-based colorimetric assay for control (C1-C3) and FRDA (P1-P5) fibroblasts grown under high-iron conditions (100 µM of FAC), with or without 100 µM of 2BP, for 16 hours. (B) Biotin immunoblots correlating with extent of DMT1 and ZIP14 palmitoylation in control (C1-C3) and FRDA (P1-P5) fibroblasts grown in regular medium. For each condition, the topmost panel shows palmitoylated DMT1 or ZIP14 levels (immunoprecipitated [IP]: biotin), the panel below shows the IP amount of DMT1 or ZIP14 (IP), and IP DMT1 or ZIP14 were used as a loading control. Immunoblotting quantifications of palmitoylated and steady-state levels of DMT1 and ZIP14 are given in supplemental Figure 7A-B. (C-F) Iron quantification for fibroblasts grown under high-iron (13 mM of holo-Tf [C-D] or +100 µM of FAC [E-F]) conditions for 72 hours, with or without 25 µM of CoA or 5 mM of DCA supplementation (C,E) or for 48 hours, with or without 25 µM of artesunate supplementation (D,F). Plots show mean ± standard error (n = 3). Two-way analyses of variance were used to compare untreated with treated values. *P < .05, ***P < .001. ns, nonsignificant.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/15/10.1182_blood.2020006987/1/m_bloodbld2020006987f6.png?Expires=1769164871&Signature=pzIfSsqORDHphHYmBDHWIah3F~mge4fThp89U18Y82AgX0SlrFQo6KxEB9kjVL3WsNCYAH9y2bgdya~HhaZW-wlLcLpVkVtAkiyHWaG8VbbtHRT6YzG0Ar8QuCSX1gHuJZIYzfeXT53zZqvIp30drMq9wY7AVwuz8hBjvkveXuJsuQg8~3fpn82CCnUZxVI4Xk1zs38vl8P~aazeBjqhiLObj5eRNvht3jP7uQJmmJ2xjL4SHASD1izJtxxJv-yvG2rchYzDW9O4N9tLEvnmH~B84JZyVRBVlCBNkbJ7ZVHI~DfhbeUqb0rrXKgTeolQszp2JpG1i4bSyHVH7fsNBA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Enhancement of TBI and NTBI uptake in FRDA fibroblasts. (A) Iron quantification using the ferrozine-based colorimetric assay for control (C1-C3) and FRDA (P1-P5) fibroblasts grown under high-iron conditions (100 µM of FAC), with or without 100 µM of 2BP, for 16 hours. (B) Biotin immunoblots correlating with extent of DMT1 and ZIP14 palmitoylation in control (C1-C3) and FRDA (P1-P5) fibroblasts grown in regular medium. For each condition, the topmost panel shows palmitoylated DMT1 or ZIP14 levels (immunoprecipitated [IP]: biotin), the panel below shows the IP amount of DMT1 or ZIP14 (IP), and IP DMT1 or ZIP14 were used as a loading control. Immunoblotting quantifications of palmitoylated and steady-state levels of DMT1 and ZIP14 are given in supplemental Figure 7A-B. (C-F) Iron quantification for fibroblasts grown under high-iron (13 mM of holo-Tf [C-D] or +100 µM of FAC [E-F]) conditions for 72 hours, with or without 25 µM of CoA or 5 mM of DCA supplementation (C,E) or for 48 hours, with or without 25 µM of artesunate supplementation (D,F). Plots show mean ± standard error (n = 3). Two-way analyses of variance were used to compare untreated with treated values. *P < .05, ***P < .001. ns, nonsignificant.
Enhancement of TBI and NTBI uptake in FRDA fibroblasts. (A) Iron quantification using the ferrozine-based colorimetric assay for control (C1-C3) and FRDA (P1-P5) fibroblasts grown under high-iron conditions (100 µM of FAC), with or without 100 µM of 2BP, for 16 hours. (B) Biotin immunoblots correlating with extent of DMT1 and ZIP14 palmitoylation in control (C1-C3) and FRDA (P1-P5) fibroblasts grown in regular medium. For each condition, the topmost panel shows palmitoylated DMT1 or ZIP14 levels (immunoprecipitated [IP]: biotin), the panel below shows the IP amount of DMT1 or ZIP14 (IP), and IP DMT1 or ZIP14 were used as a loading control. Immunoblotting quantifications of palmitoylated and steady-state levels of DMT1 and ZIP14 are given in supplemental Figure 7A-B. (C-F) Iron quantification for fibroblasts grown under high-iron (13 mM of holo-Tf [C-D] or +100 µM of FAC [E-F]) conditions for 72 hours, with or without 25 µM of CoA or 5 mM of DCA supplementation (C,E) or for 48 hours, with or without 25 µM of artesunate supplementation (D,F). Plots show mean ± standard error (n = 3). Two-way analyses of variance were used to compare untreated with treated values. *P < .05, ***P < .001. ns, nonsignificant.
Artesunate reduces iron overload in PBMCs of FRDA patients
We finally assessed whether other cell types might exhibit iron import dysregulation in FRDA. Total cellular iron content was quantified for FRDA and control PBMCs grown in FAC medium for 40 hours. After 24 hours, iron content was much higher in FRDA PBMCs, and iron overload was more pronounced after 40 hours (Figure 7A), pointing to the inability of PBMCs to regulate iron uptake under high-iron conditions. PBMC iron content in 3 heterozygote carriers was similar to that in controls, even after 40 hours of incubation. Adding 25 µM of artesunate to the PBMC culture medium resulted in a comparable decrease in iron content for both FRDA and control cells (Figure 7B). In summary, we found that regulation of iron import was impaired in FRDA PBMCs, and this characteristic could be useful for monitoring future FRDA-related clinical trials.
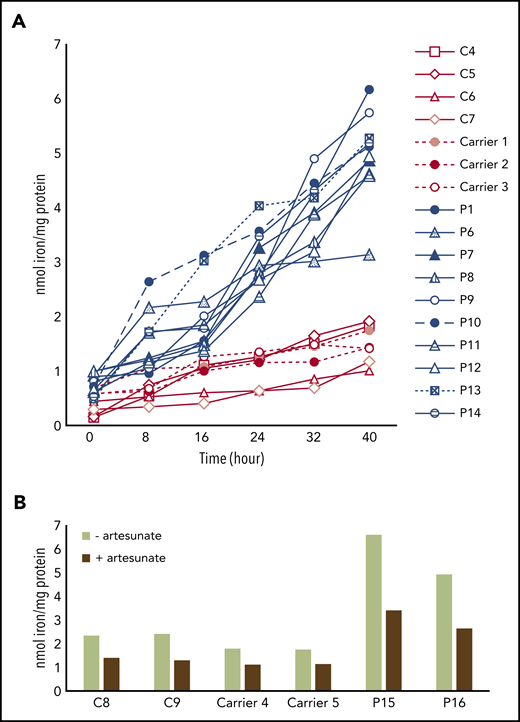
Iron overload in FRDA PBMCs limited by artesunate supplementation. (A) Iron content (at 8-hour intervals), determined by ferrozine-based colorimetric assay, of PBMCs from healthy donors (C4-C7), heterozygous carriers of FXN GAA expansions (carriers 1-3), and FRDA patients (P1, P6-P14) grown in high-iron medium (100 µM of FAC) for 40 hours (n = 1). (B) Iron content of PBMCs from controls (C8-9), heterozygous carriers of FXN GAA expansions (carriers 4-5), and FRDA patients with compound heterozygous FXN GAA expansions (P15-16) grown for 40 hours in high-iron medium (100 µM of FAC), with or without 25 µM artesunate (n = 1).
Iron overload in FRDA PBMCs limited by artesunate supplementation. (A) Iron content (at 8-hour intervals), determined by ferrozine-based colorimetric assay, of PBMCs from healthy donors (C4-C7), heterozygous carriers of FXN GAA expansions (carriers 1-3), and FRDA patients (P1, P6-P14) grown in high-iron medium (100 µM of FAC) for 40 hours (n = 1). (B) Iron content of PBMCs from controls (C8-9), heterozygous carriers of FXN GAA expansions (carriers 4-5), and FRDA patients with compound heterozygous FXN GAA expansions (P15-16) grown for 40 hours in high-iron medium (100 µM of FAC), with or without 25 µM artesunate (n = 1).
Discussion
Here we report dramatic alteration of iron homeostasis resulting from impaired regulation of TBI import in cultured fibroblasts derived from 5 FRDA patients. NTBI uptake, an important alternative source of cellular iron, was also increased, further contributing to iron overload. Both cytosolic and mitochondrial compartments accumulated abnormally large amounts of iron, a finding consistent with overproduction of mitochondrial ROS and increased ferritin and SOD1 and SOD2 steady-state levels in FRDA fibroblasts. Previous studies have reported on mitochondrial iron overload at the expense of cytosolic iron in both Δyfh1 yeast and frataxin-depleted MCK mouse mutants.6,10 Although few have evaluated cytosolic iron content, observation of increased ferritin levels in the hearts of FRDA patients,12 and livers of MCK conditional frataxin knockout mice,17 suggests cytosolic iron accumulation in several tissues.
Studying TfR1 trafficking, we noted accumulation and greater endocytosis of membrane TfR1. This prevents FRDA fibroblasts from limiting iron uptake, fostering iron overload. Moreover, delayed Tf-TfR1 recycling is expected to increase iron release from early endosomes. Cellular iron homeostasis is known to be primarily regulated by a posttranscriptional mechanism that allows TfR1 mRNA to decrease and thereby limits iron uptake under high-iron conditions.22 IRP1-IRE binding activity also decreased in FRDA fibroblasts and was expected to contribute to the reduction of TfR1 transcripts. Conversely, IRP2-IRE binding failed to decrease and even increased in FRDA fibroblasts. This feature has been reported in frataxin-, ISCU-, FDX1-, FDX2-, and FDXR-deficient animal or cellular models and ascribed to cytosolic iron depletion.14,25-28 On the basis of our data, it is conceivable that elevated IRE-IRP2 binding activity should be regarded as the consequence of defective ISC synthesis and could contribute to cytosolic iron overload. It was recently shown that FBXL5 might be able to compete with IRE for IRP2 binding, because the same IRP2 binding pocket is involved in IRE and FBXL5 interaction, the latter involving a (2Fe-2S) cluster.36 The defective ISC synthesis in FRDA should limit the ability of FBXL5 to remove IRP2 from IRE and therefore increase IRP2 steady-state level and IRP2-IRE binding activity.
The paradoxical increase in membrane TfR1 in FRDA fibroblasts also points to another level of regulation, possibly posttranslational TfR1 regulation. TfR1 palmitoylation is known to play a role in the control of cellular iron uptake. TfR1 is posttranslationally modified by S-acylation, specifically palmitoylation, with palmitate (C16:0) being the major lipid donor to S-acylated proteins.30 Here we show that TfR1 palmitoylation was defective in FRDA fibroblasts, resulting in an accumulation of membrane TfR1. Artesunate supplementation significantly increased TfR1 palmitoylation, reduced membrane TfR1 and iron content, and eventually restored the ability of FRDA fibroblasts to regulate iron uptake.
Palmitoylation increases hydrophobicity of proteins and regulates their trafficking, stability, and subcellular distribution.37 Because several endosome recycling proteins are palmitoylated, it is possible that impaired palmitoylation of other endosomal proteins yet to be identified may alter TfR1 internalization and contribute to TBI overload. It is also conceivable that impaired palmitoylation altered NTBI uptake and promoted iron overload, because 2BP exacerbated iron accumulation in both FRDA and control fibroblasts, a hypothesis supported by the decreased palmitoylation of 2 NTBI transporters, namely DMT1 and ZIP14, in FRDA fibroblasts.
Because CoA is the sole donor of acyl for palmitic acid, via palmitoyl transferases, and both FXN depletion in mice and FXN knockdown in HeLa cells reportedly alter PDH lipoylation,16,17,32-34 we hypothesized that frataxin deficiency could secondarily limit the acetyl-CoA pool. Acetyl-CoA required for TfR1 palmitoylation is mainly generated by PDH decarboxylation of pyruvate in mitochondria.35 We noted a decrease in lipoylated PDH-E2 and CoA content, confirming that FXN mutations limited the CoA/acetyl-CoA pool in FRDA fibroblasts. Conversely, CoA or DCA supplementation of cultured cells increased TfR1 palmitoylation, reduced membrane TfR1, and rescued iron overload. Interestingly, we also observed that these compounds limited TBI and NTBI uptake. We clearly established a direct relationship between frataxin defect and reduced TfR1 palmitoylation, because overexpression of FXN in FRDA fibroblasts decreased TfR1 steady-state level and reduced iron overload. Thus, by impairing ISC biogenesis, frataxin deficiency led to a secondary reduction of the CoA/acetyl-CoA pool required for normal palmitoylation and endocytosis of TfR1. Together these results suggest that CoA/acetyl-CoA, already recognized as a central metabolic intermediate and signal transducer,35 also plays an important role in the regulation of cellular iron homeostasis by controlling the extent of TfR1 palmitoylation.
In conclusion, our study not only sheds new light on the disease mechanism in FRDA, but also paves the road for evidenced-based therapeutic trials at the actionable level of TfR1 palmitoylation using existing drugs, namely artesunate and DCA. Artesunate is used to treat malaria.38 Although its safety profile and pharmacokinetics have not been assessed in neurodegenerative diseases, several millions have received artemisinin with few adverse effects. DCA is generally well tolerated when administered to PDH-deficient patients. DCA enters the circulation rapidly, crosses the blood-brain barrier, and significantly lowers blood and cerebrospinal fluid lactate concentrations.39 Our data suggest these compounds and other drugs enhancing PDH activity, acetyl-CoA synthesis, or TfR1 palmitoylation might be therapeutic agents in FRDA, especially because iron-mediated toxicity has been shown to promote neurodegeneration in a Drosophila model of FRDA.40 Finally, because FRDA PBMCs are unable to regulate iron uptake under high-iron conditions, as seen with FRDA-iPSC cardiomyocytes,41 we believe iron content and membrane TfR1 levels in patient PBMCs are potential biomarkers for future clinical trials concerned with FRDA.
For original data, please e-mail the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank C. Ottolenghi and F. Côté for helpful scientific discussions, as well as FRDA patients and their parents for their participation. The authors are also grateful for technical assistance (live microscopy and flow cytometry) from Meriem Garfa-Traoré, Nicolas Goudin, and Olivier Pellé.
Imaging and Amnis studies were performed at the Imagine Institute and funded by the Imagine Foundation, Fondation ARC, and GR-Ex LabEx. This work was supported by the Agence Nationale de la Recherche through the Investissements d’Avenir program (ANR-10-IAHU-01), the E-Rare project GENOMIT (01GM1207), and the Association Française de l’Ataxie de Friedreich.
Authorship
Contribution: F.P. designed and contributed to most of the experiments and data analyses, under the supervision of A.R. and A.M; C.Z. carried out data acquisition and analysis of iron quantification in PBMCs; M.D. helped design and interpret the Amnis experiments; A.D. and O.H. made contributions to the discussion of results and performed critical reviews of the manuscript; and F.P., A.M., and A.R. conceived this study and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Agnès Rötig, Institut Imagine, 24 Boulevard du Montparnasse, 75015 Paris, France; e-mail: agnes.rotig@inserm.fr.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal