


Abstract
von Willebrand disease (VWD) is characterized by its heterogeneous clinical manifestation, which complicates its diagnosis and management. The clinical management of VWD has remained essentially unchanged over the last 30 years or so, using von Willebrand factor (VWF) concentrates, desmopressin, and anti–fibrinolytic agents as main tools to control bleeding. This is in contrast to hemophilia A, for which a continuous innovative path has led to novel treatment modalities. Despite current VWD management being considered effective, quality-of-life studies consistently reveal a higher than anticipated burden of VWD on patients, which is particularly true for women. Apparently, despite our perceived notion of current therapeutic efficiency, there is space for innovation with the goal of reaching superior efficacy. Developing innovative treatments for VWD is complex, especially given the heterogeneity of the disease and the multifunctional nature of VWF. In this perspective article, we describe several potential strategies that could provide the basis for future VWD treatments. These include genetic approaches, such as gene therapy using dual-vector adenoassociated virus and transcriptional silencing of mutant alleles. Furthermore, protein-based approaches to increase factor FVIII levels in VWD-type 3 or 2N patients are discussed. Finally, antibody-based options to interfere with VWF degradation (for congenital VWD-type 2A or acquired von Willebrand syndrome-type 2A) or increase endogenous VWF levels (for VWD-type 1) are presented. By highlighting these potential strategies, we hope to initiate an innovative path, which ultimately would allow us to better serve VWD patients and their specific needs.
Introduction
von Willebrand disease (VWD) is an inherited bleeding disorder resulting from quantitative or qualitative deficiencies in von Willebrand factor (VWF), a plasma multimeric glycoprotein controlling platelet adhesion and aggregation.1 Although VWD is expected to affect men and women equally, there are more women diagnosed with VWD because of gynecological-related hemostatic challenges. The real prevalence of VWD has been a matter of debate for many years, ranging from as high as 1.3%, when taking into account epidemiological studies,2 to 100 cases per million, when considering patients with clinically relevant bleeding symptoms.3
VWD is traditionally classified into 3 types. VWD-type 1 is characterized by a partial quantitative deficiency in VWF. VWD-type 2 refers to qualitative VWF defects with several subtypes (2A, 2B, 2N, and 2M) according to the patients’ functional defect. Finally, VWD-type 3, also called severe VWD, is manifested by a virtually complete deficiency in VWF.1
Bleeding symptoms are usually considered less serious than hemophilia-related symptoms. Although that may be true for mild VWD patients, specific comparative studies revealed that the bleeding profile between VWD-type 3 and severe hemophilia A is more similar than expected.4,5 Nevertheless, prophylactic treatment remains relatively rare in VWD patients.
To develop alternative treatments for managing VWD in the 21st century, it will be crucial to understand the challenges faced by VWD patients and to take into account the disease specificities.
Challenges in the diagnosis of VWD
Given the heterogeneity of the disease that is observed sometimes within the same family or even in the same patient throughout his/her lifetime, VWD has been and remains to this day a complex entity to diagnose6 and to manage, partially explaining the difference between prevalence and actually diagnosed patients. Laboratory testing of VWD outside of highly specialized centers is a challenge in itself, and the link between mild phenotypes and a VWF defect is not always straightforward. In addition, bleeding history relies on a subjective interpretation by the patients, their parents as well as the clinician. This is particularly true with regard to menstrual bleeding, a subject not easily discussed both inside and outside the family circle. Weyand and James recently noted that although 76% of men with VWD are diagnosed by 10 years of age, 50% of the women are not diagnosed by 12 years of age.7 Although this could partially be explained by a first bleeding episode at different ages, it has also been shown that the diagnostic delay following the first bleeding episode is longer in women than men.8 Delayed diagnosis may result in missed treatment opportunities and increased morbidity (both medical and psychological). It is thus important to increase knowledge and awareness of VWD among (general) clinicians and public.
VWD and quality of life
What objective tools do we rely on to define that VWD is a mild bleeding disorder in most cases? First and primary is the clinical management of patients, because many VWD patients are followed in specialized hemophilia treatment centers. The health care professionals managing patients are highly trained hematologists who have the necessary expertise to evaluate the disease burden. However, and as stated maybe provocatively by Kouides, “VWD on clinical grounds is primarily a disease of females.”9 We are now increasingly learning how sex bias may have unwillingly affected patient’s care in many areas.10-12 Whether such an issue has contributed to underestimate VWD severity in terms of physical and psychological consequences is unclear. Nevertheless, there seems to be a parallel between the increasing number of female hematologists and the greater recognition of significant VWD-associated morbidities, especially when related to women’s reproductive health.13-15
The second tool to evaluate disease severity is health-related quality of life (HR-QoL) studies, widely accepted as crucial instruments of patient-centered care.16 For VWD, a few studies have addressed this issue (1) using validated HR-QoL questionnaires, (2) using questionnaires not officially validated, or (3) through interviews. All studies have reported significantly decreased quality of life in VWD patients. The largest study (509 patients) was performed in The Netherlands using the generic Short Form (SF)-36 and reported decreased vitality (score measuring energy and fatigue) in both men and women with VWD.17 Smaller studies performed in Canada (102 patients, SF-36 questionnaire), Finland (47 patients, Medical Outcomes Study-36 questionnaire), Sweden (30 female patients, SF-36 questionnaire), Canada again (28 patients, Health Utility Index Mark-2 and Mark-3), or United Kingdom (57 female patients, general questionnaire) not only confirmed a lower quality of life for VWD patients compared with the general population but also, for studies in which both men and women were included, most reported a difference between both sexes.18-22 Studies focusing primarily on women clearly indicate how the burden of VWD is borne disproportionately by women. Heavy menstrual bleeding and its associated pain (observed in 32% to 100% of patients),23 postpartum hemorrhage (in 15% to 60% of patients),13 and hysterectomy (performed in 10% to 26% of patients)24 are common in VWD women.15,25 Qualitative interview-based studies about HR-QoL or lived experiences are even more striking with women reporting a considerable impairment in their lives.26-28 Obviously not all VWD patients are affected by severe bleeding symptoms, a fact that can mitigate some of the HR-QoL studies where only a modest impact was reported, such as in the large Dutch study. Focusing on subgroups can probably give better ideas of the situation. A perfect example is illustrated in another Dutch study focusing on VWD patients with joint bleeds.29 The authors show that 23% of patients from the Dutch VWD cohort self-reported joint bleeds. Not only is this percentage higher than anticipated, but joint bleeds were reported in VWD-type 1 as well as in VWD-type 2 or VWD-type 3, even if factor VIII (FVIII) levels exceeded 10%. Joint bleeds were associated with increased consumption of factor concentrates, joint pain, and lower HR-QoL, suggesting a “significant burden of arthropathy in VWD.”29
Another factor that can impact the results of the HR-QoL studies is the absence of disease-specific instruments.23 All the validated HR-QoL questionnaires used so far are generic instruments. A VWD-specific quality of life has been developed,30 but although it was mentioned that the questionnaire was validated in a German study (Wil-QoL) and a French study (WISH-QoL), no publications are yet available.
Altogether, these different studies illustrate the higher than anticipated burden of VWD on patients and suggest that VWD classification in types 1, 2, and 3, although crucial to understand the physiopathology of the disease is not always a good proxy for clinical severity. The actual patient’s phenotype is driven not only by the VWD phenotype but also by burden in terms of hospitalizations, surgical interventions, drug consumption, gynecological visits, sick leaves, and other objective parameters. This burden is only in part included in the bleeding score used to characterize the disease (Bleeding Assessment Tool of the International Society on Thrombosis and Hemostasis). However, this tool is cumulative and more adapted to diagnosis rather than reevaluation of the bleeding tendency. To better understand the evolution of the patient’s phenotype and associated burden, an alternative score could be designed to determine spontaneous symptomatology in the presence and absence of treatment. Undoubtedly, despite our perceived notion of therapeutic efficacy of current VWD treatments, improved management of patients is necessary to address patient’s needs in an optimal fashion.
Current treatment
Over the last 30 years, many reviews related to the clinical management of VWD have been published (see, eg, Leebeek and Eikenboom,1 Kujovich,24 Aledort,31 Kruse-Jarres and Johnsen,32 Lavin and O’Donnell,33 Lavin and O’Donnell,34 Leebeek and Atiq,35 Mannucci,36 Phua and Berntorp,37 Tosetto and Castaman38 ) and guidelines were recently published.39 Without extensively revisiting the topic, some relevant issues can be highlighted.
The most relevant message is that the toolkit for the treatment of VWD has remained essentially unchanged over these 30 years. Already in 1991, Aledort described the use of intermediate-purity factor concentrates and desmopressin as main approaches for the management of VWD, as well as adjunctive antifibrinolytic and estrogenic therapy.31 It then took until 2005 for the development of a purer plasma-derived VWF concentrate, poor in FVIII activity.40 As for the first recombinant VWF concentrate, it is only since 2015 that it became available in the United States and 2017 in Europe.41
When establishing a parallel with progress made in the clinical management of hemophilia A over the same period, the contrast is particularly striking (Figure 1). Although a parallel development was observed in terms of intermediate-purity concentrates (which contain both VWF and FVIII) and desmopressin in the 1980s, a real divergence started in the 1990s with the development of monoclonal antibody-purified and recombinant FVIII concentrates. At that time, who would have guessed that it would take another 20 years for comparable products to be available for VWD? However, research did not stand still in the hemophilia field, and although VWD management was only trying to fill the gap, nothing short of a revolution was looming on the other side of the VWF/FVIII partnership. Indeed, during the last decade, hemophilia A treatment has soared with several novel therapeutic strategies being available or in late clinical development.42-45 In particular, gene therapy is reaching maturity, and therefore, for the first time in history, a real cure for the disease is within sight.
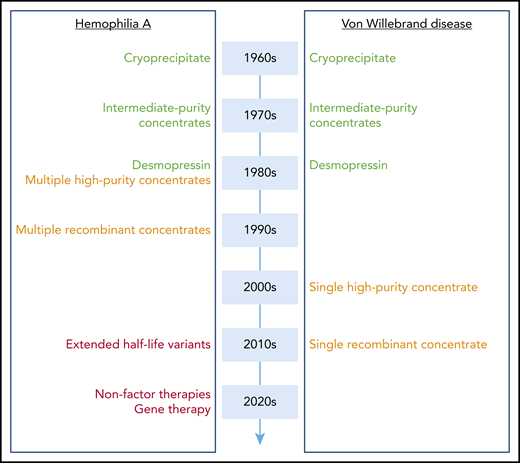
Evolution of treatment modalities for hemophilia A and VWD. A schematic overview of when novel treatment options became available for hemophilia A and VWD. High-purity and recombinant concentrates (orange) arrived 20 years later for VWD compared with hemophilia A. Extended half-life variants, nonfactor therapies, and gene therapy (red) have found their way to the clinic or are in late-stage clinical development for hemophilia A, whereas they are not yet at the horizon with regard to VWD.
Evolution of treatment modalities for hemophilia A and VWD. A schematic overview of when novel treatment options became available for hemophilia A and VWD. High-purity and recombinant concentrates (orange) arrived 20 years later for VWD compared with hemophilia A. Extended half-life variants, nonfactor therapies, and gene therapy (red) have found their way to the clinic or are in late-stage clinical development for hemophilia A, whereas they are not yet at the horizon with regard to VWD.
Thus, innovation in VWD treatment has basically stopped, and the limitations of current treatments have not been addressed. Of course, the arrival of a recombinant VWF concentrate is an important step forward. However, without neglecting its specific merits, the availability of this recombinant VWF concentrate is not changing the essentials of VWD management, and prophylactic treatment will still require multiple IV infusions per week. The invasive nature of IV administration is associated with an increased risk of infections; it is inconvenient, and it is challenging in the very young and old to find adequate venous access for repeated injections. Furthermore, prophylactic treatment using concentrates is expensive and not affordable worldwide. Finally, a major hindrance to the use of prophylaxis is that the criteria for introduction of prophylaxis for VWD patients (particularly women with heavy menstrual bleeding) are not as well defined as for patients with hemophilia. Consequently, the benefit or need for prophylaxis is less well recognized.46
The advantages of desmopressin are numerous and validate its status as the “treatment of choice” in VWD-type 1 patients. It can be administered in different ways (IV, subcutaneous, or intranasal), is relatively cheap, and is without risk of transmittable disease. It will transiently achieve adequate posttreatment levels of VWF and FVIII in 80% of cases.47,48 However, there are also some limitations. It has variable effectiveness in type 2A and 2M and is usually considered for minor bleeding/surgery.47 Moreover, this medication is contraindicated for VWD-type 2B, whereas its application in VWD-type 3 is ineffective because no FVIII or VWF is released from the endothelial storage organelles.47 Importantly, desmopressin is primarily an antidiuretic, with VWF release being an off-target effect. A number of side effects concomitant to desmopressin use have also been reported (transient headaches, facial flushing, hypotension, hyponatremia, and mild tachycardia), which are generally mild and well tolerated.49,50 Nevertheless, a recent VWD coping survey presented by Kalot et al revealed that patients/caregivers display more concern about the side-effect profile of desmopressin compared with health care professionals and seem to prefer a prophylactic approach.51 Furthermore, the repetitive use of desmopressin may result in reduced responsiveness (tachyphylaxis) due to exhaustion of the VWF storage organelles.52 Finally, it is relevant to mention that desmopressin is a monopolized product by Ferring Pharmaceuticals, which could compromise availability in the case of production issues (see, eg, https://www.ferringusa.com/press/ferring-us-issues-voluntary-nationwide-recall-of-ddavp-nasal-spray-10-mcg-0-1ml-desmopressin-acetate-nasal-spray-10-mcg-0-1ml-stimate-nasal-spray-1-5-mg-mL-due-to-superpotency/).
Hormonal treatment is commonly used to reduce heavy menstrual bleeding in VWD. However, as for desmopressin, increased VWF/FVIII levels are a mere secondary effect of hormonal treatment, which has its own side effects and contraindications. These are not always minor, as, for example, the use of progesterone has been associated with an increased risk of spheno-orbital osteomeningiomas.
In conclusion, yes, there are some efficient treatments for VWD, but, no, this does not mean that we should stop trying to improve these treatments with the goals of reaching superior efficacy, improving quality of life of the patients, and finally, curing the disease.
The challenge of innovation in VWD management
What can be done to improve VWD treatment? Unsurprisingly, there is not a simple answer to that question. Increasing prophylactic treatment would be a first step, as the introduction of prophylaxis for VWD patients appears to significantly reduce the rate of bleeding episodes and the need for hospitalization.53,54 Nevertheless, prophylactic treatment did not fully prevent the occurrence of bleeding episodes.54 In addition, the current concentrates have similar issues as the FVIII concentrates that spurred the development of novel treatment strategies for hemophilia, including a relatively short half-life requiring multiple IV infusions per week. Designing VWF variants with a prolonged half-life could be an option to reduce infusion frequency. Such a strategy would not be such a major challenge to implement, but besides an abstract published in 2006 which showed its validity, no follow-up development has been put in place.55
However, to be truly innovative, we as a scientific community need to think of alternative ways that are adapted to the patients’ situation, rather than being limited to VWF concentrates or desmopressin. It is with such a novel mindset that we will be able to break the glass ceiling and come up with the VWD-specific equivalents of fitusiran or emicizumab. In order to move in this direction, we first need to identify the patients who will better benefit from a more personalized approach, a concept increasingly recognized as relevant.37,56 As part of this identification process, we should realize that severe VWD is not necessarily restricted to VWD-type 3. Patients with VWD-type 1 or -2 may sometimes experience similar in severity bleeding phenotypes than some VWD-type 3 patients.57 Knowing the VWD subtype of the patients will be crucial to adapt treatment but should not represent a limit in our perception and appreciation of disease severity. There are patients who experience major bleeds and some who have frequent minor bleeds, and our challenge is to address the different needs of the bleeders.
In the following sections, we describe several potential approaches that could change the current paradigm of how VWD is treated or can be treated in the upcoming years (Figure 2). Of note, we do not address specifically the platelet dysfunction described in some patients with VWD-type 2B58 because targeting the VWF gene defect would also resolve the platelet phenotype.
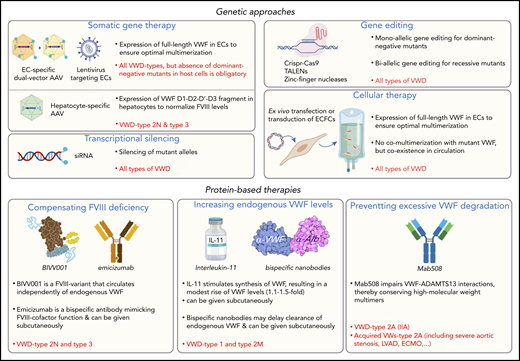
Potential avenues to enrich the arsenal of treatment options for VWD. Examples of potential novel genetic- or protein-based strategies for the management of VWD are highlighted. Some approaches could be used to serve all VWD patients, whereas others will be type specific. AAV, adeno-associated virus; ECs, endothelial cells; ECFCs, endothelial colony forming cells (also referred to as blood outgrowth endothelial cells); ECMO, extracorporal membrane oxygenation; IL-11, interleukin-11; LVAD, left ventricular assist device; TALENs, transcription activator-like effector nucleases; VWs, von Willebrand syndrome. Portions of this figure were created using Biorender.com.
Potential avenues to enrich the arsenal of treatment options for VWD. Examples of potential novel genetic- or protein-based strategies for the management of VWD are highlighted. Some approaches could be used to serve all VWD patients, whereas others will be type specific. AAV, adeno-associated virus; ECs, endothelial cells; ECFCs, endothelial colony forming cells (also referred to as blood outgrowth endothelial cells); ECMO, extracorporal membrane oxygenation; IL-11, interleukin-11; LVAD, left ventricular assist device; TALENs, transcription activator-like effector nucleases; VWs, von Willebrand syndrome. Portions of this figure were created using Biorender.com.
Novel strategies: genetic approaches
Gene therapy
To really cure VWD, gene therapy represents the goal, especially as it seems to be successful for both hemophilia A and B. However, the challenges to express VWF via gene therapy appear quite daunting. A first obstacle relates to its multimerization. Several mouse studies have shown that functional VWF can be efficiently expressed in hepatocytes of VWF-deficient mice, either transiently59-62 or for long-term expression.63,64 However, hepatocytes are not well equipped to produce sufficiently multimerized VWF, and over time the hemostatically active VWF multimers seem to disappear.63 Therefore, targeting the endothelium will be necessary for efficient protein processing.
Size represents a second hurdle: the VWF complementary DNA (cDNA) is too large to be incorporated in AAVs, the type of vectors most used in clinical trials. Successful incorporation into lentiviruses has been reported, but data on efficient in vivo expression are yet lacking.65,66
In an attempt to overcome both obstacles, our team in collaboration with Mingozzi’s team has designed a dual-AAV vector approach, in which the cDNA of VWF is divided over 2 AAV particles, which in turn are adapted to specifically target endothelial cells. We show that such an approach is technically feasible, although the current expression levels are too low to be clinically meaningful.67
However, even if we overcome these obstacles, there is another hurdle that we cannot ignore: if VWF is finally expressed at reasonable levels in the endothelial cells of the patient, how can we avoid the incorporation of dominant-negative mutants into the VWF multimer, when being expressed in the same cells as the transgene VWF? Indeed, Bowman et al reported that the inheritance pattern for VWD-type 3 proved codominant in ∼50% of the families included in their study.68
Perhaps expression of full-length VWF is not always necessary. In VWD-type 2N, and also some VWD-type 3 patients, the bleeding complications are associated with the low levels of FVIII, and increasing FVIII levels would help to reduce bleeding in joints and soft tissues. For those patients, a gene therapy approach using the N-terminal portion of VWF (D1-D2-D′-D3 domains) would result in normalization of FVIII levels, as the presence of the dimeric D′D3 fragment protects FVIII from degradation and premature clearance.69 An advantage of this approach is that multimerization is not a requirement; the cDNA (∼3.8 kb) easily fits into the AAV vector, and the protein can be expressed in hepatocytes. Preliminary studies in mice showed that such an approach is viable from a technical point of view.69
Transcriptional silencing/gene-editing
Rather than trying to express functional VWF, another approach would be to silence expression of mutant VWF. This makes even more sense given the dominant-negative character of many of the VWF mutations. A first example of this approach was reported by Casari et al, who demonstrated that small interfering RNA (siRNA)-mediated targeting of the messenger RNA breakpoint of an in-frame deletion mutant improved protein expression and multimerization.70 Recently, we applied this siRNA approach to a mouse model of VWD and confirmed the restoration of VWF multimers in plasma by silencing the dominant-negative allele.71 To find a broader application for this technology, de Jong et al designed siRNAs targeting common single-nucleotide polymorphisms in the VWF mutant allele, while leaving expression of the wild-type allele unaffected.72
Alternatively, another option could be to correct the VWF gene itself, using gene-editing approaches, such as Crispr-Cas9. Little information on this approach in view of VWD currently exists, with the exception of a study investigating the biallelic deletion of the VWF gene in ECFCs (also known as blood outgrowth endothelial cells).73
Both siRNA and gene-editing approaches are attractive, but one of the technical difficulties includes the specific targeting of sufficient numbers of endothelial cells. Currently, we do not know how many endothelial cells would need to be targeted to achieve sufficient production of functional VWF multimers. Additional research on these promising strategies is clearly needed at this point.
Cellular therapy
Ex vivo cellular therapy represents another method to introduce cells expressing functional VWF. This method was previously described by De Meyer and colleagues.65 Briefly, canine ECFCs were transduced using lentiviral vectors containing the human VWF cDNA. One day after transduction, cells indeed expressed functional multimerized VWF at levels threefold higher compared with normal ECFCs. However, expression levels decreased sevenfold over a 4-week period during the expansion phase, which could be due to in vitro culturing of these cells. Although a similar approach could also be applied using endothelial cells derived from induced pluripotent stem cells, human somatic cell therapy approaches are currently limited by the ability to generate sufficient amounts of cells to achieve physiologically relevant expression levels after transplantation, the uncertain lifespan of transplanted cells, and the occurrence of genetic changes in the cells.74 Progress of cellular therapies in other diseases will certainly be helpful for the future development of this approach regarding VWD.
Novel strategies: protein-based strategies
Compensating FVIII deficiency in VWD
Some of the bleeding complications in VWD-type 2N and type 3 patients are related to low FVIII levels, and improving FVIII levels could thus be beneficial. Infusing standard recombinant FVIII is not a viable option, because FVIII will be extremely short lived in the absence of protection conferred by VWF. On the other hand, using FVIII/VWF concentrates in type 2N patients could lead to an excess of VWF, potentially posing a thrombotic risk.75,76 An alternative solution may soon become available with the development of BIVV001, a novel FVIII-fusion variant that circulates independently of endogenous VWF, having a half-life 3 to 4 times longer than recombinant FVIII.77,78 Although being developed for hemophilia A, this molecule could provide a novel treatment option for VWD-type 2N patients to correct FVIII deficiency without increasing VWF levels.
Another approach was described by Weyand and colleagues, related to a VWD-type 3 patient who had developed alloantibodies against both FVIII and VWF and experienced recurrent bleeding episodes.79 To compensate for FVIII deficiency in this patient, treatment with emicizumab was initiated. Emicizumab is a bispecific antibody that mimics to some extent the cofactor function of FVIII.43,80 Emicizumab prophylaxis resulted in improved bleed prevention and reduced treatment burden.79
Increasing endogenous VWF levels
Desmopressin is often the first choice of treatment in VWD-type 1, as it will temporarily normalize endogenous levels of the otherwise functional VWF molecule. Because of the limitations associated with desmopressin detailed earlier, it would be of interest to develop novel strategies to increase endogenous VWF levels in a more sustainable approach. Various options could be designed in this respect. For instance, after the discovery that interleukin-11 infusion was associated with an increase in VWF levels in both mouse and canine models, its clinical benefit was evaluated in 2 phase 2 studies.81-84 Although some clinical benefit following subcutaneous application was observed, the increase in VWF levels was modest (1.1- to 1.5-fold).83,84 No additional clinical studies seem to be registered at clinicaltrials.gov.
Another strategy includes the use of single-domain antibodies (nanobodies) fused to an albumin-binding moiety (eg, peptide or nanobody), for instance, a VWF-binding nanobody that is fused to an albumin-binding peptide.85 In this approach, such fusion protein would bridge circulating VWF to albumin, taking advantage of the long albumin half-life, and induce a prolonged half-life for VWF, and consequently, increase VWF levels. Preliminary mouse studies revealed that preincubating VWF with a VWF-binding nanobody fused to an albumin-binding peptide (KB-VWF-013-ABP) resulted in a sevenfold increased VWF half-life. Moreover, subcutaneous application of this fusion protein was associated with a four- to eightfold increase in VWF and FVIII levels over a period of 7 days.85 This approach thus holds great promise but needs to be further confirmed in different preclinical models.
Preventing VWF degradation
Excessive degradation of VWF is a particular problem in congenital VWD-type 2A(IIA), but also in some variants of acquired von Willebrand syndrome, for instance, in those having severe aortic stenosis or patients requiring mechanical circulatory support. Although congenital VWD-type 2A could be treated using VWF concentrates, this is trickier in acquired von Willebrand syndrome because it seems conceivable that the infused VWF will also be susceptible to degradation while passing through the pumps. To reduce VWF degradation, antibodies can be used to interfere with ADAMTS13-VWF interactions. Importantly, inhibition of VWF degradation should be limited to avoid thrombotic side effects as seen in thrombotic thrombocytopenic purpura. A few years ago, we described an anti-VWF antibody (Mab508) targeting the ADAMTS13-docking site at the VWF-D4 domain.86 This antibody interferes with excessive VWF degradation, while leaving basal degradation intact. Indeed, the presence of Mab508 led to the conservation of high-molecular-weight multimers in ex vivo whole-blood perfusions systems.86 Given the advantages of antibody-based treatment over concentrate-based treatments (eg, long half-life, eventual subcutaneous administration), such antibodies could be valuable assets in managing patients characterized by excessive VWF degradation.
Forward-looking perspective
The heterogeneous nature of VWD implies that a “one-size-fits-all” approach will never have the potential to address all the patients’ needs. However, the management of VWD patients currently follows such a trend. As highlighted in the last few paragraphs, research has not stood still, and various solutions have been or are under investigation, although more could definitely be done in this regard. The main roadblock, in our view, appears at the next level, the translation of research results in potential industrial partnerships, allowing development of future treatments. Therefore, what are the biggest obstacles? Market size is certainly a major hurdle, with few patients likely to benefit from each of the approaches highlighted previously. Another difficulty comes from the difference in perception of disease burden between health care professionals and the patients themselves. Maybe it is time to stop comparing bleeding in hemophilia and bleeding in VWD. All HR-QoL studies tell us that VWD patients have a significantly impaired quality of life. Patient’s advocacy has therefore a critical role to play in order to bring all stakeholders around the same table in order to prepare the future. No patient should be left behind. Our main aspiration is that 10 years from now, if we were to write a review about VWD management, such a review will be quite different than the ones published for the last 30 years.
Acknowledgments
This perspective could not have been written without the input of Olivier Christophe and Caterina Casari.
This work was supported by grants from the Agence Nationale de la Recherche (ANR-17-RHUS-0011).
Authorship
Contribution: C.V.D., S.S., and P.J.L. contributed to the conception, writing, and editing of this manuscript.
Conflict-of-interest disclosure: C.V.D., S.S., and P.J.L. are inventors on patents owned by INSERM related to bleeding disorders. C.V.D., S.S., and P.J.L. are co-founders of Laelaps Therapeutics.
Correspondence: Peter J. Lenting, INSERM U1176, 80 Rue du General Leclerc, 94276 Le Kremlin-Bicêtre, France; e-mail: peter.lenting@inserm.fr.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal