

Key Points
We report a partial F8 duplication associated with markedly elevated FVIII levels and venous thrombosis in 2 Italian families.
This duplication contains transcriptional activators that may be involved in the upregulation of F8 messenger RNA and protein expression.

Abstract
High coagulation factor VIII (FVIII) levels comprise a common risk factor for venous thromboembolism (VTE), but the underlying genetic determinants are largely unknown. We investigated the molecular bases of high FVIII levels in 2 Italian families with severe thrombophilia. The proband of the first family had a history of recurrent VTE before age 50 years, with extremely and persistently elevated FVIII antigen and activity levels (>400%) as the only thrombophilic defects. Genetic analysis revealed a 23.4-kb tandem duplication of the proximal portion of the F8 gene (promoter, exon 1, and a large part of intron 1), which cosegregated with high FVIII levels in the family and was absent in 103 normal controls. Targeted screening of 50 unrelated VTE patients with FVIII levels ≥250% identified a second thrombophilic family with the same F8 rearrangement on the same genetic background, suggesting a founder effect. Carriers of the duplication from both families showed a twofold or greater upregulation of F8 messenger RNA, consistent with the presence of open chromatin signatures and enhancer elements within the duplicated region. Testing of these sequences in a luciferase reporter assay pinpointed a 927-bp region of F8 intron 1 associated with >45-fold increased reporter activity in endothelial cells, potentially mediating the F8 transcriptional enhancement observed in carriers of the duplication. In summary, we report the first thrombophilic defect in the F8 gene (designated FVIII Padua) associated with markedly elevated FVIII levels and severe thrombophilia in 2 Italian families.
Introduction
Venous thromboembolism (VTE), usually manifesting as deep vein thrombosis (DVT) and/or pulmonary embolism (PE), is a multifactorial disease affecting ∼1 in 1000 people per year. The best-known genetic factors predisposing to VTE are rare loss-of-function mutations in the natural anticoagulant proteins antithrombin,1 protein C,2 and protein S,3 as well as common gain-of-function mutations in procoagulant factors such as factor V (FV) Leiden4 and FII G20210A5 variants. More recently, sporadic gain-of-function mutations in the genes encoding coagulation FII,6-8 FV,9,10 and FIX11 have also been described in individual probands/families with severe thrombophilia. Moreover, it has been shown that elevated levels of coagulation FVIII,12 FIX,13 and FXI14 also contribute to the development of VTE. However, the genetic bases of these conditions are still poorly characterized.
FVIII, which was long believed to be produced by hepatocytes, is actually mainly synthesized in (liver sinusoid) endothelial cells15-18 and secreted in plasma as an inactive precursor. Von Willebrand factor (VWF) serves as a carrier of FVIII and prolongs its half-life in the circulation. After proteolytic activation, FVIIIa acts as an essential cofactor of FIXa in the activation of FX,19 playing a pivotal role in the amplification of blood coagulation. FVIII is encoded by the F8 gene (186 kb; 26 exons), which is located on the long arm of chromosome X (Xq28).
FVIII is the missing factor in hemophilia A, a recessive X-linked bleeding disorder affecting ∼1 in 5000 males. More than 2000 different F8 gene mutations responsible for hemophilia A have been reported, including several F8 gene rearrangements.20,21
Whereas FVIII deficiency leads to bleeding, high FVIII levels have been consistently and dose-dependently associated with an increased risk of VTE.22 In the Leiden Thrombophilia Study, it was estimated that participants with FVIII clotting (FVIII:C) ≥150%, representing ∼11% of the healthy population, had a fivefold increased risk of a first episode of VTE,12 and similar findings have been reported for recurrent VTE.22 Therefore, high FVIII levels represent a prevalent and relatively strong predisposing factor for VTE.
Elevated FVIII levels tend to cluster in families23,24 and have an estimated heritability of 30% to 60%.25-28 The best-known determinants of FVIII levels are the ABO blood group and VWF levels,29 but additional genes affecting FVIII levels have recently been identified via genome-wide association studies.30-33 In contrast, there is limited evidence for an effect of common genetic variation in the F8 gene on FVIII levels.34,35 However, it has been reported that an increased copy number of the F8 gene is associated with higher FVIII levels and VTE occurrence.36
Here, we describe a large duplication in the F8 gene associated with markedly increased FVIII levels in 2 Italian families with severe thrombophilia.
Patients and methods
Patients
Probands and family members were enrolled at Padua University Hospital and provided informed consent to participate in the study according to the Helsinki Declaration.
Blood collection and coagulation laboratory tests
Blood samples were drawn by venipuncture into vacutainer tubes containing 0.109 mol/L of sodium citrate.
Prothrombin time and activated partial thromboplastin time were determined with a BCS-XP instrument (Siemens, Milan, Italy) using Thromborel S (Siemens) and Actin FS (Siemens), respectively.
FVIII 1-stage clotting assay was performed using FVIII-deficient plasma (Siemens) and Actin FS in a BCS-XP instrument. FVIII antigen levels were assessed using the Asserachrom VIII:C Antigen Kit (Diagnostica Stago, Asnieres, France). The normal ranges obtained from 100 healthy individuals of both sexes age 20 to 70 years were 58% to 162% for the activity assay and 64% to 156% for the antigen assay. VWF antigen levels were evaluated using the HemosIL VWF:Ag Kit (Werfen, Milan, Italy) with an AcuStar instrument (Werfen). The normal range obtained from 100 healthy individuals of both sexes age 20 to 70 years was 52% to 178%.
F8 linkage analysis
Genomic DNA was isolated from peripheral blood leukocytes using the QIAamp DNA Blood Mini Kit (Qiagen). F8 linkage analysis was performed using 4 intragenic microsatellite markers. The (GT)15-20 repeat in intron 1 was amplified as described.37 Dinucleotide repeats located in introns 13 [(TG)16-24],38,39 22 [(AC)16-21],39 and 25 [(AC)13-16]40 were amplified by multiplex polymerase chain reaction (PCR). For each amplicon, 1 of the 2 primers was FAM labeled at the 5′ end (supplemental Table 1, available on the Blood Web site). After the addition of GeneScan 600 LIZ dye size standard and Hi-Di formamide (Applied Biosystems), amplification products were denatured at 80°C for 2 minutes and run on an ABI 3730 DNA Analyzer (Applied Biosystems). Peak profiles were analyzed with GeneMarker (SoftGenetics, Sate College, PA). Haplotypes were manually reconstructed taking advantage of hemizygosity in males and parent-child relationships within the families.
F8 mutation screening
The entire coding region of the F8 gene was analyzed by PCR amplification and bidirectional sequencing of individual exons using the Big Dye Terminator Sequencing Kit (version 1.1; Applied Biosystems) on an ABI Prism 3130 XL Genetic Analyzer. Sequencing chromatograms were analyzed using SeqMan Pro software (Applied Biosystems).
F8 MLPA analysis
Screening for large F8 gene rearrangements was performed by multiplex ligation-dependent probe amplification (MLPA) analysis41 using the SALSA P178 Kit (MRC-Holland, Amsterdam, The Netherlands), which contains ≥1 probes for each F8 exon and 10 reference probes targeting other genes on the X chromosome. Peak profiles obtained by capillary electrophoresis of the amplified probes were analyzed with a Coffalyser analysis tool using the default parameters. Briefly, the peak height of each F8-specific probe was normalized against the average peak height of all reference probes. A peak ratio of ∼1.0 indicates that that particular F8 exon is present in a normal copy number (ie, 1 copy in males and 2 copies in females), whereas peak ratios of ∼2.0 (in males) and ∼1.5 (in females) indicate duplication.
Droplet digital PCR
Single Color Droplet Digital PCR (QX200 workflow; Bio-Rad, Hercules, CA)42 of F8 exon 1 (target) and a distant X-linked amplicon (reference) was carried out according to the manufacturer’s instructions. Primers (supplemental Table 2) and experimental details are reported in the data supplement.
WGS
The data supplement provides detailed descriptions of the experimental protocol and analysis pipeline. Briefly, DNA libraries were prepared using the TruSeq Nano DNA HT Sample Prep Kit (Illumina, San Diego, CA) following the manufacturer’s instructions and sequenced on an Illumina HiSeq platform. Quality-controlled paired-end reads were aligned to the human reference genome (version GRCh37/hg19) using Burrows-Wheeler Aligner software.43 Structural variants were identified using CNVnator44 and LUMPY45 packages. The breakpoint of the duplication was confirmed by Sanger sequencing using the primers listed in supplemental Table 3. These same primers were used to screen the general population for carriers of the F8 duplication. The chromosome X haplotype underlying the F8 duplication was reconstructed from the whole-genome sequencing (WGS) data of hemizygous males from each family, eliminating the need for genotype phasing.
F8 mRNA analysis
Total RNA was isolated from blood mononuclear cells using TRIzol Reagent (Thermo Fisher Scientific, Waltham, MA) and an RNeasy Kit (Qiagen), including an on-column DNase digestion step. After quantification at the NanoDrop, RNA (1 µg) was reverse transcribed at 42°C for 2 hours using Superscript II enzyme (Thermo Fisher Scientific) and an oligo-d(T) primer. The obtained complementary DNA was amplified using EvaGreen Master Mix (Solis Biodyne, Tartu, Estonia) and primers located in F8 exon 22 (ie, outside the duplicated region; supplemental Table 4). F8 expression was normalized to 2 housekeeping genes (GAPDH and ACTB) using the 2−ΔΔCt method.46
In silico analysis of the duplicated region
The in silico analysis was carried out on the hg19 assembly of the Human Reference Genome. The duplicated region of the F8 gene was screened for enhancer sequences (EnhancerAtlas),47 pattern of H3K27Ac histone modification,48 and DNase I hypersensitivity clusters (Encode V3). Intron-mediated enhancement (IME) elements were identified by blasting the IME consensus sequence (TTNGATYTG)49 against the duplicated DNA region with FIMO software,50 as reported in the data supplement.
Fragment C of the duplicated region was also analyzed for the presence of transcription factor (TF) binding sites and clusters of TFs using different bioinformatic tools, as detailed in the data supplement.
Luciferase reporter assays
Putative regulatory elements identified by the in silico analysis were amplified by PCR and directionally cloned in the pGL4.26[luc2/minP/Hygro] vector (Promega, Madison, WI), upstream of a minimal promoter (ie, a TATA-box promoter element already present in the vector) driving the transcription of Firefly luciferase. Cloning primers are listed in supplemental Table 5, and cloned fragments are annotated in supplemental Table 6. The pGL4.74 [hRluc/TK] vector (Promega), containing the Renilla luciferase gene under control of an HVS-TK promoter, was used as a control in cotransfection experiments. Luciferase assays were performed in endothelial cells (human umbilical vein endothelial cells [HUVECs]) and hepatic cells (HepG2), as described in the data supplement.
Results
Family A
Proband A (member II:3 of family A; Figure 1; Table 1) presented with DVT in the right leg and PE after a cesarean section at age 31 years and was treated with low-molecular-weight heparin (LMWH) followed by warfarin for 6 months. At age 44 years, she developed idiopathic DVT in the left leg, which was treated with warfarin for 1 year without recurrence. Thrombophilic screening was unremarkable, except for extremely high FVIII levels (FVIII:C, 422%; FV:Ag, 432%), which persisted at follow-up for several years, despite normal levels of VWF. After a new idiopathic DVT in the left leg at age 46 years, the patient was put on long-term warfarin treatment, but at age 52 years, she developed recurrent DVT in the right leg and PE, despite her prothrombin time/international normalized ratio being in the therapeutic range. At this time, her FVIII level was >500%, and after hospital admission, she received therapeutic-dose LMWH. Eventually, she was diagnosed with advanced-stage uterine leiomyosarcoma and died 8 months later as a result of acute kidney failure complications.
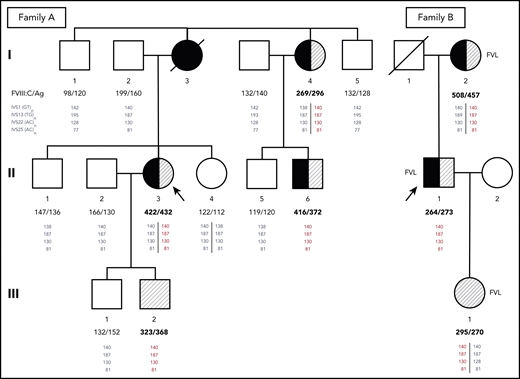
Pedigrees of families A and B. Probands are indicated by arrows. Family members who have experienced DVT and/or PE are shown as closed or half-closed symbols. Individuals with markedly elevated FVIII levels (FVIII:C >250%) are hatched, whereas carriers of the FV Leiden mutation (FVL) are indicated next to the symbols of family B. FVIII:C and FVIII antigen (FVIII:Ag) levels expressed in percentages are reported below each individual. Phased genotypes for the microsatellite markers located in introns (IVS) 1, 13, 22, and 25 of the F8 gene are also reported below each individual. Microsatellite alleles are identified by the respective PCR product sizes in bp. The haplotype shared by all family members with markedly elevated FVIII levels (140-187-130-81) is shown in red. Note that although the duplication is always associated with this haplotype, the reverse is not true, and several family members have inherited copies of the same haplotype without the duplication (shown in gray as all other haplotypes that are not associated with the duplication).
Pedigrees of families A and B. Probands are indicated by arrows. Family members who have experienced DVT and/or PE are shown as closed or half-closed symbols. Individuals with markedly elevated FVIII levels (FVIII:C >250%) are hatched, whereas carriers of the FV Leiden mutation (FVL) are indicated next to the symbols of family B. FVIII:C and FVIII antigen (FVIII:Ag) levels expressed in percentages are reported below each individual. Phased genotypes for the microsatellite markers located in introns (IVS) 1, 13, 22, and 25 of the F8 gene are also reported below each individual. Microsatellite alleles are identified by the respective PCR product sizes in bp. The haplotype shared by all family members with markedly elevated FVIII levels (140-187-130-81) is shown in red. Note that although the duplication is always associated with this haplotype, the reverse is not true, and several family members have inherited copies of the same haplotype without the duplication (shown in gray as all other haplotypes that are not associated with the duplication).
Clinical characteristics and laboratory data of the family members
| Participant | Sex | Age, y | Thrombotic events | Age at first VTE, y | PT/INR | aPTT, s* | FVIII:C, %† | FVIII:Ag, %‡ | VWF:Ag, %§ | Additional thrombophilic defects |
|---|---|---|---|---|---|---|---|---|---|---|
| Family A | ||||||||||
| II-3, proband | F | 53‖ | DVT and PE | 31 | 2.88¶ | 26.5¶ | 422 | 432 | 165 | No |
| I-4 | F | 77 | DVT and PE | 49 | 1.00 | 21.6 | 269 | 296 | 144 | No |
| II-6 | M | 45 | SVT and DVT | 43 | 0.98 | 21.1 | 416 | 372 | 60 | No |
| III-1 | M | 26 | No | — | 1.11 | 29.3 | 132 | 152 | 144 | No |
| III-2 | M | 22 | No | — | 1.1 | 27.2 | 323 | 368 | 112 | No |
| Family B | ||||||||||
| II-1, proband | M | 41 | DVT and PE | 21 | 3.1¶ | 40¶ | 264 | 273 | 78 | Heterozygous FV Leiden |
| I-2 | F | 66 | DVT | 24 | 0.93 | 22.2 | 508 | 457 | 68 | Heterozygous FV Leiden |
| III-1 | F | 19 | No | — | 1.07 | 26.2 | 295 | 270 | 113 | Heterozygous FV Leiden |
| Participant | Sex | Age, y | Thrombotic events | Age at first VTE, y | PT/INR | aPTT, s* | FVIII:C, %† | FVIII:Ag, %‡ | VWF:Ag, %§ | Additional thrombophilic defects |
|---|---|---|---|---|---|---|---|---|---|---|
| Family A | ||||||||||
| II-3, proband | F | 53‖ | DVT and PE | 31 | 2.88¶ | 26.5¶ | 422 | 432 | 165 | No |
| I-4 | F | 77 | DVT and PE | 49 | 1.00 | 21.6 | 269 | 296 | 144 | No |
| II-6 | M | 45 | SVT and DVT | 43 | 0.98 | 21.1 | 416 | 372 | 60 | No |
| III-1 | M | 26 | No | — | 1.11 | 29.3 | 132 | 152 | 144 | No |
| III-2 | M | 22 | No | — | 1.1 | 27.2 | 323 | 368 | 112 | No |
| Family B | ||||||||||
| II-1, proband | M | 41 | DVT and PE | 21 | 3.1¶ | 40¶ | 264 | 273 | 78 | Heterozygous FV Leiden |
| I-2 | F | 66 | DVT | 24 | 0.93 | 22.2 | 508 | 457 | 68 | Heterozygous FV Leiden |
| III-1 | F | 19 | No | — | 1.07 | 26.2 | 295 | 270 | 113 | Heterozygous FV Leiden |
Participants are coded as in Figure 1.
INR, international normalized ratio; PT, prothrombin time; SVT, superficial vein thrombosis.
Normal range, 26 to 34 seconds.
Normal range, 58% to 162%.
Normal range, 64% to 156%.
Normal range, 52% to 178%.
Age at death.
Oral anticoagulant treatment (warfarin).
Among the proband’s relatives, the mother (I:3) had died as a result of PE at age 48 years after a leg fracture and immobilization, whereas her father (I:2), a heavy smoker with hypertension and dyslipidemia, had experienced myocardial infarction and right retinal artery thrombosis at age 68 years. A maternal aunt (I:4) had suffered from DVT in her left leg and PE at age 49 years after immobilization for twisting her left knee and recently developed saphenous vein thrombosis in the left leg that was treated with fondaparinux. Because of atrial fibrillation, she was currently receiving anticoagulation with apixaban. One cousin (II:6) on the maternal side presented with unprovoked saphenous vein thrombosis and common femoral DVT in the right leg at age 43 years. Thrombophilic screening in family members revealed markedly elevated FVIII levels in the proband’s maternal aunt (I:4; FVIII:C, 269%; FVIII:Ag, 296%) and cousin (II:6; FVIII:C, 416%; FVIII:Ag, 372%) as well as in the proband’s asymptomatic younger son (III:2; FVIII:C, 323%; FVIII:Ag, 368%) in the presence of normal VWF levels. The main clinical characteristics and laboratory data of family A are summarized in Table 1.
Genetic analysis in family A
The distribution of high FVIII levels in the family was compatible with X-linked dominant inheritance, pointing to F8 as an obvious candidate gene. Involvement of the F8 gene was further supported by a linkage analysis with intragenic microsatellite markers, revealing a shared F8 gene haplotype (140-187-130-81) in all family members with high FVIII levels (Figure 1). However, no mutation was found by sequencing the proximal promoter51 (∼1000 bp) and all exons and splicing junctions of the F8 gene.
To check for possible amplification of the F8 gene, MLPA analysis was carried out in male and female family members with high (III:2 and II:3) or normal (III:1 and II:4) FVIII levels. This analysis revealed the presence of 1 extra copy of exon 1 in the family members with high FVIII levels (Figure 2). Extension of the MLPA analysis to other relatives showed that all family members with high FVIII levels were hemizigous (males) or heterozygous (females) for the F8 exon 1 duplication, whereas none of the family members with normal FVIII levels carried this rearrangement (data not shown).
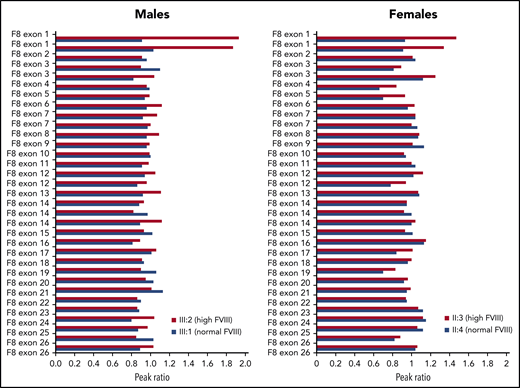
MLPA analysis of the F8 gene in 4 members of family A.F8 MLPA profiles are shown for 2 males with high and normal FVIII levels, respectively (left) and 2 females with high and normal FVIII levels, respectively (right), coded as in Figure 1. The peak obtained for each F8-specific probe was normalized against the average of the peaks of the reference probes in the same DNA sample, and the peak ratio was plotted as a function of the corresponding F8 exon. A peak ratio of ∼1.0 indicates a normal number of copies (1 for males, 2 for females), whereas peak ratios of ∼2.0 (in males) or ∼1.5 (in females) indicate a duplication of the sequence recognized by that probe. Note that some F8 exons, including exon 1, are targeted by >1 probe.
MLPA analysis of the F8 gene in 4 members of family A.F8 MLPA profiles are shown for 2 males with high and normal FVIII levels, respectively (left) and 2 females with high and normal FVIII levels, respectively (right), coded as in Figure 1. The peak obtained for each F8-specific probe was normalized against the average of the peaks of the reference probes in the same DNA sample, and the peak ratio was plotted as a function of the corresponding F8 exon. A peak ratio of ∼1.0 indicates a normal number of copies (1 for males, 2 for females), whereas peak ratios of ∼2.0 (in males) or ∼1.5 (in females) indicate a duplication of the sequence recognized by that probe. Note that some F8 exons, including exon 1, are targeted by >1 probe.
The duplication was confirmed by droplet digital PCR (supplemental Figure 1) and mapped by WGS in a male member of the family (III:2). Analysis of the sequencing data showed that a large genomic region spanning the proximal portion of the F8 gene had higher coverage (ie, was overrepresented in the sequencing reads) than the surrounding sequence, roughly defining the extension of the duplication (Figure 3A). The identification of reads simultaneously mapping to the F8 promoter and to the distal portion of intron 1, which were several kb apart in the reference sequence, allowed us to define the genomic arrangement of the duplication and define its boundaries at the nucleotide level. The duplication spanned 23 420 bp (GRCh37, 154 229 849-154 253 268) and included the promoter, exon 1, and nearly the whole intron 1 of the F8 gene. The 2 copies were arranged in tandem without spacer DNA and had the same orientation (Figure 3B). This genomic arrangement and the exact junction point were confirmed by Sanger sequencing (Figure 3B). Both breakpoints of the duplication fell within a repetitive element: an L2 long interspersed nuclear element for the upstream boundary in the promoter region and an AluJo short interspersed nuclear element and L1MB2 long interspersed nuclear element for the downstream boundary in intron 1. Moreover, close inspection of the junction point sequence revealed that the 2 breakpoints of the duplication shared an identical stretch of 7 nucleotides (Figure 3B inset), suggesting that the duplication had arisen by a microhomology-mediated mechanism.52 No additional insertions or deletions were observed at either end of the duplicated region.
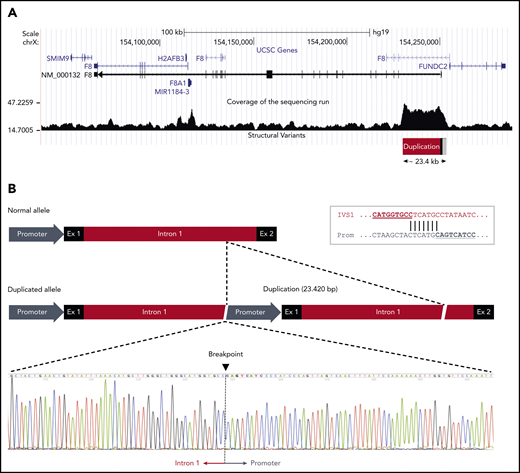
F8 duplication and breakpoint mapping in family A. (A) Genomic region spanning the F8 gene according to the University of California Santa Cruz Genome Browser structure. The sequencing coverage in member III:2 of family A is plotted at the bottom. The duplicated region of ∼23.4 kb at the 5′ end of the F8 transcript (NM_000132) is indicated. Note that the F8 gene is oriented from right to left in this scheme. (B) Comparison of the normal and duplicated alleles (oriented from left to right). The exact breakpoint of the duplication, where intron 1 of the upstream copy is joined to the promoter region of the downstream copy, is indicated in the sequencing chromatogram. The inset in the top right corner shows the 7-bp sequence of microhomology between intron 1 and promoter that might have mediated the duplication event.
F8 duplication and breakpoint mapping in family A. (A) Genomic region spanning the F8 gene according to the University of California Santa Cruz Genome Browser structure. The sequencing coverage in member III:2 of family A is plotted at the bottom. The duplicated region of ∼23.4 kb at the 5′ end of the F8 transcript (NM_000132) is indicated. Note that the F8 gene is oriented from right to left in this scheme. (B) Comparison of the normal and duplicated alleles (oriented from left to right). The exact breakpoint of the duplication, where intron 1 of the upstream copy is joined to the promoter region of the downstream copy, is indicated in the sequencing chromatogram. The inset in the top right corner shows the 7-bp sequence of microhomology between intron 1 and promoter that might have mediated the duplication event.
Identification of another family with the same F8 duplication (family B)
PCR-based screening of 103 individuals with normal FVIII levels (52 males and 51 females, for a total of 154 F8 alleles) from the same geographical area failed to detect any carriers of the same F8 duplication, excluding the possibility of a polymorphic copy-number variation. The duplication was also absent from the 1000 Genome project database.53
However, MLPA-based screening of 50 unrelated VTE patients with FVIII:C ≥250%, in the absence of acquired conditions known to increase FVIII levels (eg, inflammation, corticosteroid treatment, Cushing’s syndrome, liver cirrhosis, or infection) revealed a second patient with exactly the same F8 duplication (as confirmed by WGS). This 41-year-old patient (proband B; member II;1 of family B; Figure 1; Table 1) who carried the FV Leiden mutation in addition to having elevated FVIII levels (FVIII:Ag, 273%; FVIII:C, 264%), had experienced a first episode of unprovoked DVT and PE at age 21 years. He was treated with LMWH and warfarin for 1 year. At age 31 years, he experienced a new episode of unprovoked PE. After the initial treatment with LMWH, lifelong warfarin therapy was recommended, and no recurrences had occurred until now (age 41 years). His 66-year-old mother (FVIII:Ag, 457%; FVIII:C, 508%) and 19-year-old daughter (FVIII:Ag, 270%; FVIII:C, 295%) also carried the F8 duplication, as well as FV Leiden. His mother had experienced popliteal DVT in the right leg 20 days after cesarean section at age 24 years. She recently developed saphenous vein thrombosis in the right leg. The proband’s daughter remained still asymptomatic. All 3 family members had normal VWF levels. The main clinical characteristics and laboratory data of family B are summarized in Table 1.
Genotyping of the microsatellite markers located in F8 introns 1, 13, 22, and 25 showed that the F8 haplotype underlying the duplication in this family (140-187-130-81) was exactly the same as in family A (Figure 1). Further analysis of the single-nucleotide polymorphisms surrounding the duplicated region in male carriers from both families (III:2 and II:1, respectively) revealed an unusually long (∼3 Mb) shared haplotype underlying the duplication in both families (supplemental Table 7), indicating a shared origin of the F8 duplication in a common ancestor who lived at least 13 generations (∼350 years) ago (details provided in data supplement under “Mapping of reads to the reference sequence and relatedness analyses”).
F8 mRNA analysis
To gain more insight into the mechanisms by which the identified F8 duplication may increase FVIII levels, F8 messenger RNA (mRNA) expression in blood lymphocytes was quantified by quantitative reverse transcription polymerase chain reaction. This analysis showed that F8 mRNA expression was upregulated two- to eightfold in carriers of the F8 duplication from both families compared with normal controls (Figure 4), suggesting that the duplication increases the transcription of the F8 gene.
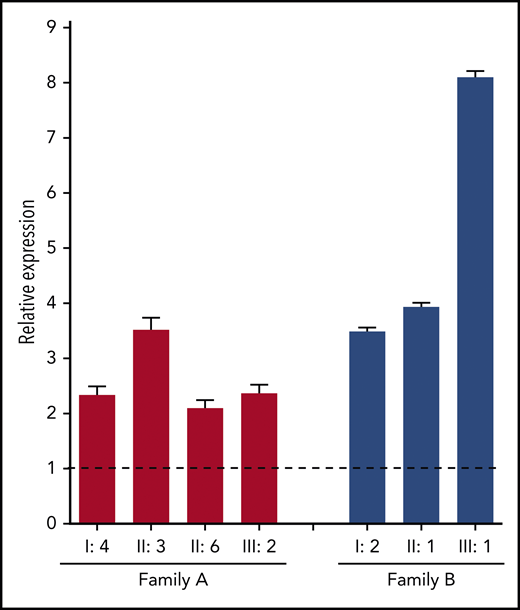
F8 mRNA analysis. Total RNA from peripheral blood lymphocytes was reverse transcribed with an oligo-(dT) primer, and F8 exon 22 was amplified in carriers of the F8 duplication from both families (coded as in Figure 1) and in 4 normal controls. The relative expression of F8 mRNA in carriers of the F8 duplication vs normal controls was calculated using ACTB and GAPDH as reference genes. Error bars represent standard deviations of at least 3 determinations. The dashed line represents the normal F8 expression level in controls.
F8 mRNA analysis. Total RNA from peripheral blood lymphocytes was reverse transcribed with an oligo-(dT) primer, and F8 exon 22 was amplified in carriers of the F8 duplication from both families (coded as in Figure 1) and in 4 normal controls. The relative expression of F8 mRNA in carriers of the F8 duplication vs normal controls was calculated using ACTB and GAPDH as reference genes. Error bars represent standard deviations of at least 3 determinations. The dashed line represents the normal F8 expression level in controls.
Functional analysis of the F8 duplication
In an attempt to explain the F8 transcriptional enhancement observed in carriers of the duplication, the duplicated region was examined in silico for the presence of chromatin features indicative of active transcription (DNase I hypersensitivity regions, H3K27Ac histone modification) and for regulatory elements such as TF binding sites, enhancers, and IME sequences (Figure 5A). This analysis resulted in the identification of 2 main genomic regions potentially enhancing the transcription of the F8 gene. The first region (chr X, 154 250 176-154 252 008), denominated region F, corresponded to the whole F8 promoter (with binding sites for several TFs), exon 1, and the proximal portion of intron 1 and contained the highest H3K27Ac peaks at the 5′ end of the F8 gene. The second region (chr X, 154 232 683-154 240 123), entirely located within intron 1, spanned several regulatory elements and was therefore subdivided into 5 smaller fragments (regions A-E) according to the positions of TF binding sites and DNase I hypersensitivity clusters from ENCODE (Figure 5A). The genomic coordinates and sequences of regions A to F are detailed in supplemental Tables 5 and 6.
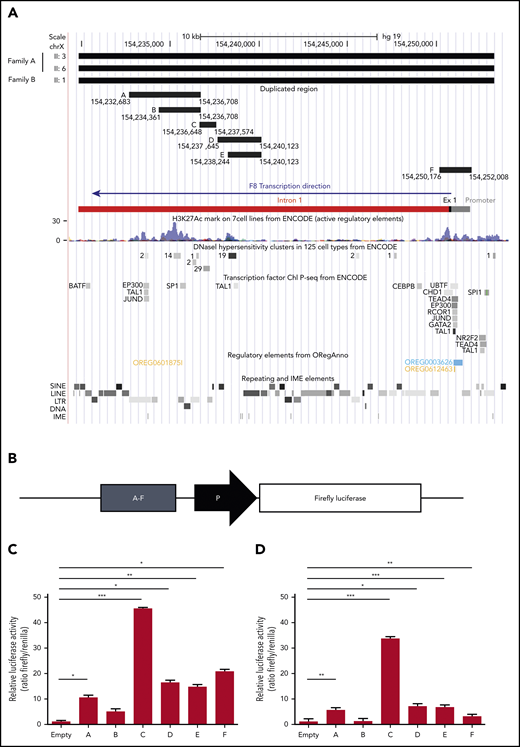
Functional characterization of the duplicated region. (A) In silico identification of regulatory elements in the duplicated DNA region. The duplicated region identified by WGS of 3 patients from both families is shown at the top. The genomic coordinates of the putative enhancer regions A, B, C, D, E, and F (supplemental Tables 5 and 6) are indicated. The F8 proximal promoter, exon 1, and intron 1, color coded as in Figure 3, are shown for additional reference. The locations of H3K27Ac peaks, DNase I hypersensitivity sites, and binding sites for specific TFs (from ENCODE), as well as repeating and regulatory elements (extracted from ORegAnno) and IME motifs (identified with FIMO), are illustrated in the genome browser tracks below the F8 gene bar. (B) Regions A to F were cloned (1 by 1) in a reporter vector upstream of a minimal promoter (ie, a TATA-box promoter element already present in the empty vector) driving Firefly luciferase expression and transfected into HUVECs and HepG2 cells together with a vector expressing Renilla luciferase. (C-D) After 12 hours, cells were lysed, and luciferase activities were determined in HUVECs (C) and HepG2 cells (D). Data are normalized for empty vector (containing only the minimal promoter) and Renilla fluorescence. The bars represent means and standard deviations of 6 (HUVECs) or 4 (HepG2 cells) replicates. *P < .05, **P < .001, ***P < .0001. LINE, long interspersed nuclear element; LTR, long terminal repeat; SINE, short interspersed nuclear element.
Functional characterization of the duplicated region. (A) In silico identification of regulatory elements in the duplicated DNA region. The duplicated region identified by WGS of 3 patients from both families is shown at the top. The genomic coordinates of the putative enhancer regions A, B, C, D, E, and F (supplemental Tables 5 and 6) are indicated. The F8 proximal promoter, exon 1, and intron 1, color coded as in Figure 3, are shown for additional reference. The locations of H3K27Ac peaks, DNase I hypersensitivity sites, and binding sites for specific TFs (from ENCODE), as well as repeating and regulatory elements (extracted from ORegAnno) and IME motifs (identified with FIMO), are illustrated in the genome browser tracks below the F8 gene bar. (B) Regions A to F were cloned (1 by 1) in a reporter vector upstream of a minimal promoter (ie, a TATA-box promoter element already present in the empty vector) driving Firefly luciferase expression and transfected into HUVECs and HepG2 cells together with a vector expressing Renilla luciferase. (C-D) After 12 hours, cells were lysed, and luciferase activities were determined in HUVECs (C) and HepG2 cells (D). Data are normalized for empty vector (containing only the minimal promoter) and Renilla fluorescence. The bars represent means and standard deviations of 6 (HUVECs) or 4 (HepG2 cells) replicates. *P < .05, **P < .001, ***P < .0001. LINE, long interspersed nuclear element; LTR, long terminal repeat; SINE, short interspersed nuclear element.
The transcriptional activity of regions A to F was quantified in luciferase reporter assays. To this end, each region was cloned in a reporter vector upstream of a minimal promoter driving the expression of Firefly luciferase (Figure 5B) and transfected into endothelial cells (HUVECs) together with a control plasmid-expressing Renilla luciferase. Quantification of luciferase activity 12 hours posttransfection showed that all regions except B significantly increased Firefly luciferase expression (Figure 5C). By far the highest transcriptional activity (>45-fold enhancement) was associated with region C, followed by region F (∼20-fold enhancement), which contained the F8 proximal promoter. Regions D and E were equally active, despite the presence of an additional H3K27Ac domain in region D, whereas the activity of region A compared with the largely overlapping region B could be associated with a regulatory element binding EP300, TAL1, and JunD, which was absent in B.
Essentially the same results, but slightly lower signals, were obtained in HepG2 cells (Figure 5D). Only region F was clearly more active in HUVECs than in HepG2 cells, in line with a recent in vivo study demonstrating higher activity of the human F8 promoter in mouse liver sinusoidal endothelial cells compared with hepatocytes.18 Region C, enhancing luciferase activity >30-fold, was again the most potent.
Region C (GRCh37, 154 236 648-154 237 574) overlapped a major DNase I hypersensitivity cluster in various cell types (Figure 5A) and was predicted to bind to several general TFs (eg, SP1 and STATs; supplemental Figure 2) as well as members of the ETS family (supplemental Figure 3), which have recently been shown to play important roles in the physiological expression of the F8 gene in endothelial cells.18,54 Interestingly, the ETS binding site was found to cluster with binding sites for other TFs that play roles in endothelial pathophysiology, such as the TEF family,55 LSF (also known as TFCP2),56 and AP-1,57 all located on the same negative DNA strand as the F8 gene itself (supplemental Figure 3).
Discussion
Elevated FVIII levels constitute a well-established risk factor for VTE,22 but their genetic determinants remain largely elusive. Here we describe a tandem duplication of the proximal portion of the F8 gene associated with extremely high FVIII levels in 2 thrombophilic families. The causative role of this F8 duplication is supported by several lines of evidence, including: its perfect cosegregation with FVIII levels >250% in 2 different families, its absence in 103 normal controls (154 alleles) from the same geographical area, the associated twofold or greater increase in F8 mRNA in blood cells, the presence of numerous transcription control elements within the duplicated region, and the ability of some of these elements to increase the expression of a reporter gene when cloned upstream of a minimal promoter. Moreover, our findings fit well with the emerging role of structural variants, and particularly duplications, as determinants of gene expression outliers.58
Although it was not possible to analyze the F8 mRNA in liver sinusoidal endothelial cells, the upregulation of F8 mRNA expression observed in blood cells of carriers of the duplication pointed to transcriptional enhancement as the most likely mechanism underlying the increased FVIII levels. Because the duplication included the whole proximal promoter region51 (Figure 3B), transcription of the duplicated allele could in principle be driven by both promoters. Although the availability of 2 equivalent transcription start sites served by independent promoters could by itself increase the overall transcription rate, the transcription efficiency of the downstream promoter may be further enhanced by the presence of regulatory sequences that reside in intron 1 and are placed upstream of this promoter by the duplication. In fact, the leading intron of genes is known to be enriched in transcriptional enhancers and chromatin marks that promote gene expression,59-61 and several portions of intron 1 actually enhanced luciferase expression in reporter assays. In particular, region C, containing a major DNase I hypersensitivity cluster and several TF binding sites, increased luciferase activity >45-fold in HUVECs and >30-fold in HepG2 cells. Interestingly, this region contained a cluster of binding sites for several (endothelial) TFs, including ETS, the role of which in the transcriptional regulation of F8 has recently been demonstrated.54
Although our reporter constructs could not recapitulate the full in vivo complexity of the duplication, which may involve positional and epigenetic effects as well, they do provide evidence for the presence of cis-acting elements within the duplicated region. Additional studies are needed to fully characterize the transcriptionally active regions and determine whether the duplication acts by enhancing the physiological FVIII expression in endothelial cells or possibly also by upregulating ectopic expression in other cell types (eg, hepatocytes).62 Moreover, in the light of the multiple transcription start sites and complex 5′ alternative processing of F8 transcripts,63 it remains to be determined whether the mRNA produced by the duplicated allele is qualitatively normal. In fact, qualitative alterations of the mRNA, particularly of the 5′ untranslated region, might contribute to increased FVIII expression by influencing mRNA stability and translation.
The observed variation in FVIII levels in carriers of the F8 duplication may be due to age, acquired conditions, genetic variation in other genes, and skewed X inactivation in females. This is clinically relevant, considering that VTE risk increases with FVIII levels in a dose-dependent manner.22 Moreover, the F8 duplication may synergize with other risk factors for VTE, as in family B, where the penetrance of the F8 duplication was likely increased by coinheritance of the FV Leiden mutation. Notably, in both families, 5 of 7 carriers of the duplication developed (recurrent) VTE at a young age.
Although large F8 gene rearrangements are a relatively common cause of hemophilia A,20,21 our F8 duplication is the first example of a rearrangement associated with thrombophilia rather than hemophilia. Interestingly, Shen et al36 previously observed that the F8 gene is often amplified in Chinese VTE patients and that F8 copy number correlates with FVIII levels. Because their assessment of F8 copy number was based exclusively on quantitative PCR of an F8 exon 1–intron 1 amplicon, it is possible that at least some of their patients actually carried partial duplications of the proximal portion of the F8 gene (similar to that described here) rather than whole-gene duplications. In fact, F8 intron 1 is a well-known breakpoint and rearrangement hotspot in hemophilia A patients,21,64-68 and a number of duplications involving exon 1, but not associated with hemophilia A, have been reported as incidental findings.69 In our case, exactly the same F8 duplication was present in 2 European patients from the same geographical area (northeastern Italy), who had inherited it from a remote common ancestor, suggesting a founder effect. Population screening for this particular duplication can be performed by PCR amplification of the duplication breakpoint using primers F8_dupl_BP_F-R (supplemental Table 3), which yielded an 844-bp product in carriers of the duplication and no product in noncarriers.
In summary, we have identified a gain-of-function tandem duplication of the proximal portion of the F8 gene associated with markedly elevated FVIII levels and VTE. Additional studies are needed to fully elucidate the exact molecular mechanisms by which this rearrangement increases FVIII levels and determine whether this is a unique case or the first example of a more general mechanism of familial thrombophilia.
For original data, please email the corresponding author at paolo.simioni@unipd.it.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank S. Spierts for her assistance with the GeneMarker software.
This study was supported by funds from the Department of Medicine, University of Padua Medical School (P.S.), and the Cardiovascular Research Institute Maastricht (E. Castoldi), as well as by the European Union through European Regional Development Fund Project #2014-2020.4.01.16-0024 MOBTT53 (L.P.).
Authorship
Contribution: P.S. identified the probands and families; P.S., D.T., L.S., and E. Campello were responsible for patient management, selection, and enrollment; S.G., F.S., C.M.R., and C.B. performed coagulation laboratory workup; C.B. performed direct sequencing of the F8 gene; E. Castoldi and F.N. were responsible for linkage analysis, identification, and preliminary mapping of the duplication; F.S. performed duplication confirmation and screening; S.C., F.S., and F.C. performed WGS, quantitative PCR, and luciferase assays; G.S., L.P., and S.C. were responsible for bioinformatics; E. Castoldi, S.C., and P.S. integrated and interpreted the data; E. Castoldi, P.S., and S.C. were responsible for manuscript writing and figures; T.M.H. critically reviewed the manuscript; and P.S. (clinical and coagulation laboratory), E. Castoldi (molecular genetics), and S.C. (functional genomics) supervised the study.
Conflict-of-interest disclosure: T.M.H. is a shareholder in Coagulation Profile BV (Maastricht, The Netherlands). The remaining authors declare no competing financial interests.
Correspondence: Paolo Simioni, General Internal Medicine, Thrombotic and Hemorrhagic Disorder Unit, Department of Medicine, University of Padua Medical School, Via Giustiniani 2, Padua, Italy; e-mail: paolo.simioni@unipd.it.
