

Key Points
Heme inhibits human plasma B-cell differentiation by blocking the DOCK8/STAT3 signaling pathway and by modulating HO-1 enzymatic activity.
B cells from alloimmunized SCD patients are resistant to heme inhibition but resistance can be reversed with cotreatment with quinine.

Abstract
Red blood cell alloimmunization remains a barrier for safe and effective transfusions in sickle cell disease (SCD), but the associated risk factors remain largely unknown. Intravascular hemolysis, a hallmark of SCD, results in the release of heme with potent immunomodulatory activity, although its effect on SCD humoral response, specifically alloimmunization, remains unclear. Here, we found that cell-free heme suppresses human B-cell plasmablast and plasma cell differentiation by inhibiting the DOCK8/STAT3 signaling pathway, which is critical for B-cell activation, as well as by upregulating heme oxygenase 1 (HO-1) through its enzymatic byproducts, carbon monoxide and biliverdin. Whereas nonalloimmunized SCD B cells were inhibited by exogenous heme, B cells from the alloimmunized group were nonresponsive to heme inhibition and readily differentiated into plasma cells. Consistent with a differential B-cell response to hemolysis, we found elevated B-cell basal levels of DOCK8 and higher HO-1–mediated inhibition of activated B cells in nonalloimmunized compared with alloimmunized SCD patients. To overcome the alloimmunized B-cell heme insensitivity, we screened several heme-binding molecules and identified quinine as a potent inhibitor of B-cell activity, reversing the resistance to heme suppression in alloimmunized patients. B-cell inhibition by quinine occurred only in the presence of heme and through HO-1 induction. Altogether, these data suggest that hemolysis can dampen the humoral B-cell response and that B-cell heme responsiveness maybe a determinant of alloimmunization risk in SCD. By restoring B-cell heme sensitivity, quinine may have therapeutic potential to prevent and inhibit alloimmunization in SCD patients.
Introduction
Red blood cell (RBC) alloimmunization remains a major barrier to safe transfusions. Patients with sickle cell disease (SCD), for whom transfusions remain a cornerstone treatment,1,2 are at highest risk of alloimmunization,3,4 with potentially life-threatening outcomes.3,5-7 Genetic as well as acquired patient-related factors are likely to influence the process of alloimmunization. Although genome-wide association studies have identified potential genetic associations with the alloantibody responder phenotype,8,9 none to date have been validated as an alloimmunization biomarker.
Hemolysis is a hallmark of SCD.10,11 When sickle hemoglobin is deoxygenated, it polymerizes, causing changes in RBC membrane shape and function that increase its fragility and ultimately lead to RBC destruction and hemoglobin release. Increasing evidence suggests that hemoglobin and its oxidized form of free heme, a lipophilic bioactive molecule,12 play a key role in the initiation and progression of hemolytic complications because of their ability to trigger oxidative stress,13-15 sterile inflammation,16-18 cell death,19,20 and tissue injury.21 With regard to alloimmunization, we have previously described differential innate immune control of T-cell skewing between nonalloimmunized and alloimmunized SCD patients under hemolytic conditions, in part because of differences in monocyte levels of heme oxygenase 1 (HO-1),22 an immunoregulatory enzyme with anticytotoxic, and anti-inflammatory properties.23 In addition, differences in heme-mediated NF-κB activation and maturation of T helper type 1 polarizing dendritic cells were found between alloimmunized and nonalloimmunized SCD patients.24 However, direct effects of hemolysis on human humoral immune cell response and RBC alloimmunization remain largely unknown. With respect to the effects on B cells, a study in mice showed that heme increases plasma B-cell differentiation and immunoglobulin M (IgM) production through binding and induction of Bach2 degradation.25 In addition, mitochondrial derived reactive oxygen species was shown to inhibit mouse plasma cell differentiation by reducing endogenous heme synthesis.26 However, the effect of heme on human B cells has not yet been studied. One report found that Src-Syk-Stat3 activation through DOCK8, an adaptor protein that binds to free heme, is important for B-cell activation and function,27 although the role of heme or hemolysis was not examined. Interestingly, a deficiency in DOCK8 is associated with impairment of memory B-cell and marginal zone B-cell development.27-30 We therefore reasoned that free heme, through binding to DOCK8, may inhibit B-cell activation and that differential heme signaling in B cells via STAT3 may dictate whether humoral immunity against transfused cells is aborted (nonalloimmunized) or induced (alloimmunized).
Here, we examined the effect of hemolysis on human B-cell development and tested the hypothesis that B-cell response to hemolysis may differ between alloimmunized and nonalloimmunized SCD patients.
Materials and methods
All studies were approved by the Institutional Review Boards of the New York Blood Center and Montefiore Health Center. Detailed experimental procedures are outlined in supplemental Data (available on the Blood Web site). Briefly, naïve or memory B cells were isolated from peripheral blood of healthy donors (HDs) and SCD patients and were stimulated in the absence or presence of hemin and/or various inhibitors for 7 days followed by analysis of B-cell proliferation, plasmablast or plasma cell differentiation, and DOCK8 and HO-1 expression using flow cytometry. Naïve B cells were stimulated for 30 minutes for protein phosphorylation analysis using phospho-flow cytometry. Basal DOCK8 and HO-1 levels in circulating B cells before stimulation were analyzed by flow cytometry. Levels of total heme and related hemolytic parameters in SCD plasma were measured using commercially available kits.
Results
Hemolysis inhibits plasmablast cell differentiation
To test the effect of free heme on human B-cell activation, purified circulating naive (CD27–) and memory B cells (CD27+) from HDs labeled with CFSE were subjected to polyclonal activation to induce antigen-independent differentiation into antibody-secreting cells in the absence or presence of cell-free heme. After 7 days, the extent of proliferated (CFSElo) CD19+ B cells and plasmablast differentiation (CD19loCD38hi), including IgG+ isotype switched B cells (CD19loCD38hiIgG+) within live B cells (supplemental Figure 1A), was measured by flow cytometry (Figure 1A; supplemental Figure 1C). On the basis of our previous study in monocytes,22 we tested the effect of 2.5 μM heme first. In contrast to a minor inhibition of proliferation, exogenous heme had a robust inhibitory effect on differentiation of naïve B cells into plasmablasts (P = .0016; Figure 1B). The inhibitory effect was dose-dependent, with 6.8% ± 1.3% inhibition of B-cell proliferation and 52.7% ± 8.0% inhibition of plasmablast cell differentiation at the highest concentration tested (5 μM) (Figure 1C). Interesting, heme had no effect on memory B-cell proliferation or differentiation (supplemental Figure 1B). Similarly, B-cell class switch recombination, measured as percent IgG+ B cells in proliferated B cells and plasmablast cells, was not affected by heme (supplemental Figure 1C-D). We should note that all our B-cell analyses were restricted to live cells only and that even at the highest dose of heme used, percentages of viable B cells were high (supplemental Figure 1G).
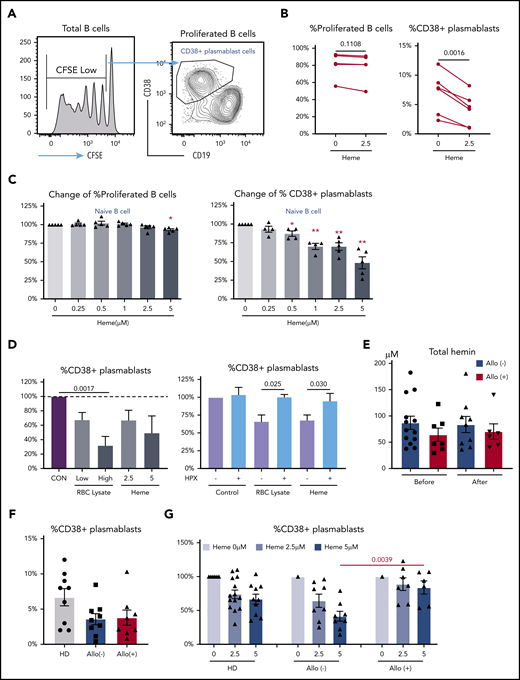
Heme inhibits B-cell activation. Purified naïve B cells from HDs were CFSE labeled and stimulated with B-cell activation cocktail for 7 days. (A-B) By using the gating strategy for analysis of proliferated B cells and CD38+ plasmablasts (A), we showed the frequency of proliferated B cells in total B cells and of CD38+ plasmablasts in the absence or presence of 2.5 μM heme (B). (C) Fold change in proliferated B-cell and CD38+ plasmablast frequencies in the presence of increasing doses of heme relative to no heme treatment. (D) Fold change in CD38+ plasmablast frequencies in the presence of different doses of RBC lysate or heme (left) or a given RBC lysate dose or 2.5 μM heme preincubated or not with 2.5 μM hemopexin (HPX; right). (E) Cell-free heme levels just before transfusion or within 4 hours after transfusion in plasma from allo-positive and allo-negative SCD patients. (F) CD38+ plasmablast frequencies in stimulated naïve B cells from HDs and allo-positive and allo-negative SCD patients. (G) Fold change in CD38+ plasmablast frequencies in the presence of 2.5 μM or 5 μM heme. Data represent means ± standard error of the mean. *P < .05; **P < .01. CON, control.
Heme inhibits B-cell activation. Purified naïve B cells from HDs were CFSE labeled and stimulated with B-cell activation cocktail for 7 days. (A-B) By using the gating strategy for analysis of proliferated B cells and CD38+ plasmablasts (A), we showed the frequency of proliferated B cells in total B cells and of CD38+ plasmablasts in the absence or presence of 2.5 μM heme (B). (C) Fold change in proliferated B-cell and CD38+ plasmablast frequencies in the presence of increasing doses of heme relative to no heme treatment. (D) Fold change in CD38+ plasmablast frequencies in the presence of different doses of RBC lysate or heme (left) or a given RBC lysate dose or 2.5 μM heme preincubated or not with 2.5 μM hemopexin (HPX; right). (E) Cell-free heme levels just before transfusion or within 4 hours after transfusion in plasma from allo-positive and allo-negative SCD patients. (F) CD38+ plasmablast frequencies in stimulated naïve B cells from HDs and allo-positive and allo-negative SCD patients. (G) Fold change in CD38+ plasmablast frequencies in the presence of 2.5 μM or 5 μM heme. Data represent means ± standard error of the mean. *P < .05; **P < .01. CON, control.
RBC lysates also induced a similar inhibition of plasmablast differentiation (P = .0017; Figure 1D). Preincubation of RBC lysate (1 g/L hemoglobin31 ) or free heme (2.5 μM) with hemopexin (a heme scavenging protein; 2.5 μM) reversed the inhibitory effect (Figure 1D), further confirming that inhibition was specific to heme. Altogether, these data suggest that plasma-free heme can inhibit naïve B-cell activation predominantly at the plasmablast B-cell differentiation stage.
Altered B-cell heme response in alloimmunized patients with SCD
We reasoned that the inhibitory effect of heme on B-cell activation may modulate the risk of alloimmunization in hemolytic disorders such as SCD. As a first step to test this, we compared levels of intravascular hemolysis, specifically total plasma heme levels, between alloimmunized (allo-positive) and nonalloimmunized (allo-negative) patients with SCD who received transfusions, but we did not find any significant differences either before or after transfusion (Figure 1E), suggesting that intravascular heme levels do not correlate with the alloimmunized or nonalloimmunized state of an SCD patient. Similarly, no differences were found in any hemolytic indicator such as sickle hemoglobin percentage, reticulocyte percentage, lactate dehydrogenase, hemopexin, haptoglobin, or bilirubin levels (supplemental Table 1), and differentiation of HD-naïve B cells into plasmablasts was comparable in the presence of sera from allo-negative or allo-positive SCD patients (supplemental Figure 1E). We next tested whether B-cell responses to cell-free heme differed in these 2 groups of patients. In the absence of heme, naïve B cells from allo-negative and allo-positive SCD patients differentiated comparably into plasmablasts (Figure 1F). However, in the presence of heme, B cells from allo-negative patients were more sensitive to the effects of heme than B cells from allo-positive SCD patients (Figure 1G), with 58.6% ± 7.3% inhibition of plasmablast B-cell differentiation in allo-negative SCD patients but only 16% ± 9.7% in allo-positive SCD patients at the highest heme (5 μM) concentration (allo-negative vs allo-positive P = .0039). Consistent with the minor effect of heme on HD B-cell proliferative responses (Figure 1B), the effect of heme on B-cell proliferation did not differ between allo-negative and allo-positive patients (supplemental Figure 1F). These data indicate that allo-negative, but not allo-positive, SCD B cells are sensitive to the effects of heme, and that their differentiation into plasma cells is inhibited by hemolysis, raising the possibility that this inhibitory effect of heme may lower the risk of RBC alloimmunization, but only in the allo-negative SCD group.
Cell-free heme inhibits plasma cell differentiation through the DOCK8 signaling pathway
It has been shown that plasma-free heme can directly bind to DOCK8 protein and inhibit monocyte or macrophage phagocytosis through the DOCK8-Cdc42 signaling pathway.32 DOCK8/STAT3 signaling pathway activation, leading to phosphorylation of Pyk2, SYK, SRC, and STAT3, has also been reported to be critical for B-cell activation,27 although the effect of heme on this activation pathway was not examined. To test whether DOCK8/STAT3 signaling is inhibited by cell-free heme during B-cell activation, we analyzed the phosphorylation of Pyk2, SYK, SRC, and STAT3 after short-term stimulation in the presence or absence of heme (10 μM) by intracellular flow cytometric analysis. In the absence of heme, stimulation of HD B cells induced phosphorylation of Pyk2, SYK, SRC, and STAT3 (Figure 2A). Heme treatment resulted in significant reduction in the levels of phosphorylated Pyk2 (pPyk2), pSYK, pSRC, and pSTAT3 (Figure 2B) (P = .0088, .0063, .0091, and .0313, respectively). The inhibitory effect of heme on pSTAT3 was confirmed in overnight samples (supplemental Figure 2B). Levels of pPyk2, pSRC, and pSYK were decreased by 60 minutes and were not analyzed after that time point (supplemental Figure 2A). These data indicate heme-mediated reduction of several key phosphoproteins in the DOCK8/STAT3 signaling pathway in B cells, consistent with inhibition of B-cell DOCK8/STAT3 activation by heme.
![Inhibition of B-cell activation by heme is through the DOCK8/STAT3 signaling pathway. (A) Purified HD naïve B cells were stimulated with B-cell activation cocktail, and phosphorylation of Pyk2 (pPyk2), pSyk, pSRC, and pSTAT3 was analyzed by intracellular flow cytometry after 15 minutes. Histogram overlays depict the extent of phosphorylation without (gray) or with stimulation (blue) with the phosphorylated signal gate placed by reference to the baseline prestimulation histogram. (B) Purified HD naïve B cells were stimulated for the same length of time as in panel A in the absence or presence of heme (10 μM), and the frequency of cells positive for pPyk2, pSyk, pSRC, and pSTAT3 is shown. (C) Purified HD naïve B cells were stimulated with B-cell activation cocktail for 7 days. Contour plots depict gating strategy for DOCK8 Low and DOCK8 High in proliferated B cells as well as plasma B cells (CD27hiBlimp1+ cells) within DOCK8low or DOCK8high cells. (D) Fold change in plasma B-cell frequencies in 7-day purified HD naïve B-cell–stimulated cultures in the presence of 2.5 μM or 5 μM heme relative to no heme stimulated B cells. (E) DOCK8high (left) and DOCK8low (right) cell numbers in stimulated B-cell cultures treated with 2.5 μM or 5 μM heme. Fold change in DOCK8high or DOCK8low cell numbers with heme treatment relative to no treatment. (F) Representative histogram overlay showing DOCK8 expression (blue) in peripheral B cells relative to isotype control (gray). Relative DOCK8 expression (mean fluorescence intensity [MFI]) in circulating B cells from HD and allo-negative and allo-positive patients is shown. (G) Fold change in DOCK8high B-cell and CD27hi plasma cell frequency in 7-day heme-treated cultures of stimulated naïve B cells from allo-negative and allo-positive patients. Data represent means ± standard error of the mean. SSC, side scatter (light).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/2/10.1182_blood.2020008511/4/m_bloodbld2020008511f2.png?Expires=1769085586&Signature=IqWW-zC8k0pc9L9IsXWaQOg6MFJCpfW0bZk0HAoasPRr49wH7F~VIpLYSVapm5RwBjffvpKRquRowdCTZ~1wpKvCQb~txNQrhmJ~oJJwxb4dKK~XIOIfdRB5-sT2VMaYFHXk4EG70rBADbhQktpC5CQpuF6SbPPWq80TCdU0eRThzb2xnjcAIGdA5vArdc9x8UOrkiUzG8mIII559-avw6wNBt3iol5v8Gn4pkxpKsQqi4SVbLxEKC1nC7Imw7UjU35UKbmUmvnhV2hoBLKTHdH6eQjO~rka-COPSE7sIGrUzHfXBmSZpFhcXSO7E7v1iaesZiJ4BH1kPVX64jtEAg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Inhibition of B-cell activation by heme is through the DOCK8/STAT3 signaling pathway. (A) Purified HD naïve B cells were stimulated with B-cell activation cocktail, and phosphorylation of Pyk2 (pPyk2), pSyk, pSRC, and pSTAT3 was analyzed by intracellular flow cytometry after 15 minutes. Histogram overlays depict the extent of phosphorylation without (gray) or with stimulation (blue) with the phosphorylated signal gate placed by reference to the baseline prestimulation histogram. (B) Purified HD naïve B cells were stimulated for the same length of time as in panel A in the absence or presence of heme (10 μM), and the frequency of cells positive for pPyk2, pSyk, pSRC, and pSTAT3 is shown. (C) Purified HD naïve B cells were stimulated with B-cell activation cocktail for 7 days. Contour plots depict gating strategy for DOCK8 Low and DOCK8 High in proliferated B cells as well as plasma B cells (CD27hiBlimp1+ cells) within DOCK8low or DOCK8high cells. (D) Fold change in plasma B-cell frequencies in 7-day purified HD naïve B-cell–stimulated cultures in the presence of 2.5 μM or 5 μM heme relative to no heme stimulated B cells. (E) DOCK8high (left) and DOCK8low (right) cell numbers in stimulated B-cell cultures treated with 2.5 μM or 5 μM heme. Fold change in DOCK8high or DOCK8low cell numbers with heme treatment relative to no treatment. (F) Representative histogram overlay showing DOCK8 expression (blue) in peripheral B cells relative to isotype control (gray). Relative DOCK8 expression (mean fluorescence intensity [MFI]) in circulating B cells from HD and allo-negative and allo-positive patients is shown. (G) Fold change in DOCK8high B-cell and CD27hi plasma cell frequency in 7-day heme-treated cultures of stimulated naïve B cells from allo-negative and allo-positive patients. Data represent means ± standard error of the mean. SSC, side scatter (light).
Inhibition of B-cell activation by heme is through the DOCK8/STAT3 signaling pathway. (A) Purified HD naïve B cells were stimulated with B-cell activation cocktail, and phosphorylation of Pyk2 (pPyk2), pSyk, pSRC, and pSTAT3 was analyzed by intracellular flow cytometry after 15 minutes. Histogram overlays depict the extent of phosphorylation without (gray) or with stimulation (blue) with the phosphorylated signal gate placed by reference to the baseline prestimulation histogram. (B) Purified HD naïve B cells were stimulated for the same length of time as in panel A in the absence or presence of heme (10 μM), and the frequency of cells positive for pPyk2, pSyk, pSRC, and pSTAT3 is shown. (C) Purified HD naïve B cells were stimulated with B-cell activation cocktail for 7 days. Contour plots depict gating strategy for DOCK8 Low and DOCK8 High in proliferated B cells as well as plasma B cells (CD27hiBlimp1+ cells) within DOCK8low or DOCK8high cells. (D) Fold change in plasma B-cell frequencies in 7-day purified HD naïve B-cell–stimulated cultures in the presence of 2.5 μM or 5 μM heme relative to no heme stimulated B cells. (E) DOCK8high (left) and DOCK8low (right) cell numbers in stimulated B-cell cultures treated with 2.5 μM or 5 μM heme. Fold change in DOCK8high or DOCK8low cell numbers with heme treatment relative to no treatment. (F) Representative histogram overlay showing DOCK8 expression (blue) in peripheral B cells relative to isotype control (gray). Relative DOCK8 expression (mean fluorescence intensity [MFI]) in circulating B cells from HD and allo-negative and allo-positive patients is shown. (G) Fold change in DOCK8high B-cell and CD27hi plasma cell frequency in 7-day heme-treated cultures of stimulated naïve B cells from allo-negative and allo-positive patients. Data represent means ± standard error of the mean. SSC, side scatter (light).
We next tested the effect of cell-free heme on DOCK8 signaling during B-cell differentiation. Strikingly, we found that stimulated B cells could be divided into DOCK8 high (DOCK8high) and DOCK8 low (DOCK8low) cell subsets based on DOCK8 expression (Figure 2C, left). By using CD27 and Blimp-1 as plasma cell markers,33 we found that CD27hiBlimp-1+ plasma cells were mostly within the DOCK8high rather than the DOCK8low subset (Figure 2C, right). Heme treatment led to inhibition of CD27hiBlimp-1+ plasma cell differentiation (P = .0195; Figure 2D). We also found a dose-dependent decrease of DOCK8-expressing cells in stimulated B cells, with a more robust reduction in DOCK8high than DOCK8low subsets (61.6% ± 6.5% vs 34.1% ± 5.9% at 5 μM; P = .0084; Figure 2E), suggesting that DOCK8high cells are preferentially inhibited by heme and that heme inhibits plasma cell differentiation by targeting DOCK8high- activated B cells.
To test whether DOCK8 expression is associated with differential heme responses, we measured intracellular DOCK8 expression in circulating B cells, and as controls, in T cells and monocytes. Almost all B cells were DOCK8+ as previously reported,34 but DOCK8 levels were significantly higher in allo-negative compared with allo-positive B cells (P < .0001; Figure 2F), but not in T cells or monocytes (supplemental Figure 2C). Importantly, heme inhibited DOCK8high B cells and CD27hiBlimp-1+ plasma cell differentiation in allo-negative SCD patients, but not in allo-positive patients (Figure 2G), suggesting that DOCK8 expression levels may be associated with heme response in B cells and SCD alloimmunization risk.
Heme inhibits B-cell activation through modulation of HO-1 enzyme activity
The inhibitory effect of heme on B-cell activation may involve signaling pathways other than just DOCK8/STAT3. HO-1 is a key enzyme for heme degradation with multiple biologic activities attributed to its enzymatic byproducts.23 We analyzed HO-1 expression levels in human B cells before and after stimulation with our polyclonal B-cell activation cocktail. HO-1 expression was low in B cells before stimulation (Figure 3A, left side), but increased significantly after stimulation, mostly in CD38+ plasmablasts (Figure 3A, right side) and was further induced by treatment with heme (Figure 3B). We also found higher HO-1 levels in DOCK8hi plasma cells (supplemental Figure 3B).
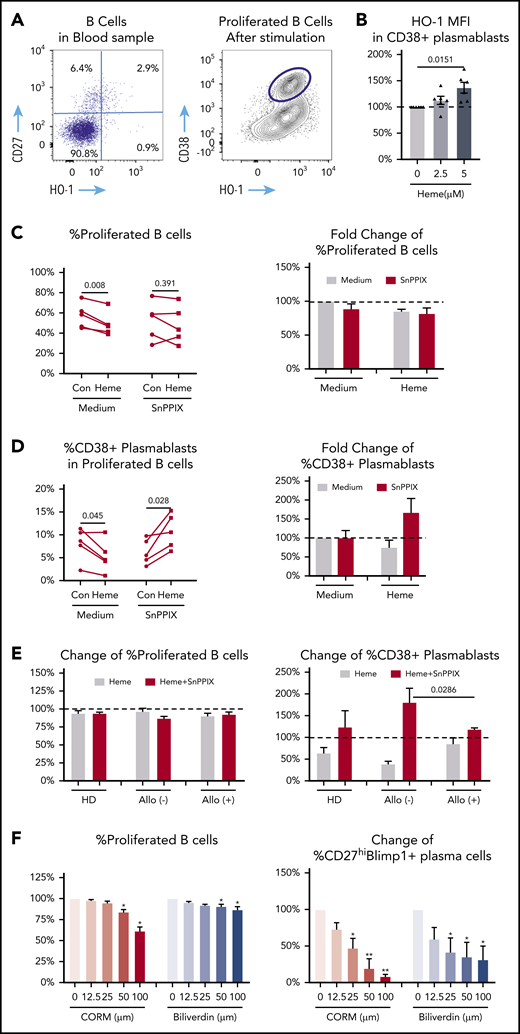
Role of HO-1 enzyme activity in heme-mediated B-cell activation. (A) HO-1 analysis of HD unstimulated peripheral blood B cells (left) and after 7-day stimulation of purified naïve B cells in proliferated B cells (gate showing CD38+ plasmablasts). (B) Fold change in HO-1 expression in plasma cells in 7-day stimulated purified HD B cells treated with 2.5 μM or 5 μM heme relative to no heme treatment. (C-D) Absolute and fold change of frequency of B-cell proliferation (C) and CD38+ plasmablasts in the presence of SnPPIX (2.5 μM) without or with (2.5 μM) heme (D). (E) Fold change in B-cell proliferation and CD38+ plasmablasts 7-day stimulated naïve B cells from allo-positive and allo-negative SCD patients in the absence or presence of SnPPIX (2.5 μM) and heme (2.5 μM) relative to untreated media control (Con). (F) Fold change in B-cell proliferation and plasma B cells in 7-day stimulated HD naïve B cells in the presence of different doses of heme degradation byproducts, carbon monoxide releasing molecule 3 (CORM-3) and biliverdin, relative to untreated cultures. Data represent means ± standard error of the mean. *P < .05; **P < .01.
Role of HO-1 enzyme activity in heme-mediated B-cell activation. (A) HO-1 analysis of HD unstimulated peripheral blood B cells (left) and after 7-day stimulation of purified naïve B cells in proliferated B cells (gate showing CD38+ plasmablasts). (B) Fold change in HO-1 expression in plasma cells in 7-day stimulated purified HD B cells treated with 2.5 μM or 5 μM heme relative to no heme treatment. (C-D) Absolute and fold change of frequency of B-cell proliferation (C) and CD38+ plasmablasts in the presence of SnPPIX (2.5 μM) without or with (2.5 μM) heme (D). (E) Fold change in B-cell proliferation and CD38+ plasmablasts 7-day stimulated naïve B cells from allo-positive and allo-negative SCD patients in the absence or presence of SnPPIX (2.5 μM) and heme (2.5 μM) relative to untreated media control (Con). (F) Fold change in B-cell proliferation and plasma B cells in 7-day stimulated HD naïve B cells in the presence of different doses of heme degradation byproducts, carbon monoxide releasing molecule 3 (CORM-3) and biliverdin, relative to untreated cultures. Data represent means ± standard error of the mean. *P < .05; **P < .01.
To examine the role of HO-1 enzymatic activity in heme-mediated inhibition of B-cell activation, B cells were stimulated in the presence of heme without or with SnPPIX (2.5 μM), a competitive HO-1 inhibitor that blocks its activity. Pretreatment with heme or SnPPIX alone or heme plus SnPPIX had little to no effect on B-cell proliferation (Figure 3C), indicating that HO-1 was largely devoid of pro- or antiproliferative activity. In contrast, heme-mediated inhibition of plasmablast cell differentiation was reversed by SnPPIX, and treatment with heme plus SnPPIX led to an even higher frequency of plasmablasts compared with no treatment (Figure 3D), suggesting that inability to degrade heme may induce plasma cell differentiation by other as yet unknown pathways. SnPPIX alone had no effect on differentiation (8.1% ± 3.5% vs 6.3% ± 2.7%; P > .3; Figure 3D), indicating that basal intracellular heme levels are too low to provide enough substrate for HO-1 enzymatic activity to inhibit B-cell differentiation. We also tested the effect of SnPPIX on SCD allo-positive and allo-negative B-cell activation. B-cell proliferative responses were comparable in the presence of SnPPIX in the 2 groups (Figure 3E). In contrast, SnPPIX reversed heme-mediated inhibition of plasmablast differentiation in allo-negative SCD patients but had less effect in allo-positive SCD patients (Figure 3E), suggesting a more profound inhibitory effect of HO-1 in B cells from allo-negative SCD patients. Altogether, these data suggest that HO-1 enzymatic activity is involved in abetting decreased plasmablast B-cell differentiation by heme and that HO-1–mediated inhibition of activated B cells is more pronounced in allo-negative than allo-positive SCD patients.
HO-1 can degrade heme into carbon monoxide (CO), iron, and biliverdin.23 Iron chelation at high concentrations inhibited both B-cell proliferation and differentiation (data not shown), whereas, it had inconsistent effects at low concentrations (supplemental Figure 3). In contrast, treatment with CO releasing molecule-3 (CORM-3) and exogenous biliverdin inhibited B-cell proliferation and plasma cell differentiation in a dose-dependent manner with more robust effects on plasma cell differentiation than B-cell proliferation (CO: 91.2% ± 2.9% vs 38.7% ± 5.0%; biliverdin: 68.4% ± 18.6% vs 13.3% ± 3.5% at the highest concentrations; Figure 3F). These data are consistent with a role for the HO-1 enzymatic byproducts CO and biliverdin in the inhibition of plasma B-cell differentiation.
Quinine enhances the heme inhibitory effect on B-cell activation in both allo-negative and allo-positive SCD patients
Several small molecule-based antimalarial drugs have been developed on the basis of their ability to bind and alter heme bioactivity.35-38 We reasoned that some of these molecules may also alter the B-cell inhibitory properties of heme and may, in some cases, further bolster the inhibitory effects of heme and even reverse refractoriness to heme-mediated B-cell inhibition as seen in SCD allo-positive patients. To test this, several heme-binding molecules including quinine (QA), chloroquine (CQ), amodiaquine (AQ), and dihydroartemisinin (DHA) were tested on HD B-cell responses. AQ and CQ inhibited plasma cell differentiation in the absence or presence of heme, whereas the inhibitory effect of QA and DHA on B cells required the presence of heme with a more robust inhibition by QA than DHA (Figure 4A). QA inhibition was dose-dependent, and at higher doses of heme and QA (2.5 μM), we found an almost complete inhibition of plasma cell differentiation (Figure 4B), consistent with an additive effect on suppression of B-cell activation. Of note, although all analyses were restricted to live cells, viability studies indicated minimal cytotoxic effects of QA except at the highest doses of heme plus QA (supplemental Figure 4B).
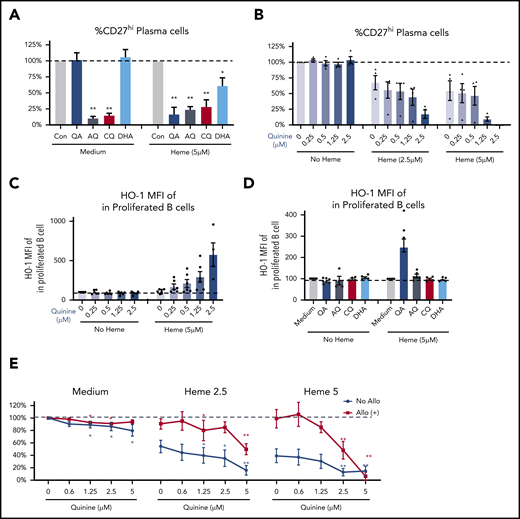
QA increases heme-mediated inhibition of B-cell activation. (A-B) Fold change in plasma cell frequency in 7-day stimulated purified naïve HD B cells in the presence of 2.5 μM QA, AQ, CQ, or DHA without or with 5 μM heme relative to untreated but stimulated cultures (A), or different doses of QA plus 2.5 μM or 5 μM heme (B). (C) Levels of HO-1 expression (MFI) in proliferated B cells of 7-day stimulated B cells treated with different doses of QA without or with 5 μM heme. (D) Fold change in HO-1 expression in stimulated B cells in the presence of 2.5 μM QA, AQ, CQ, or DHA without or with 5 μM free heme relative to untreated but stimulated cultures. (E) Fold change in plasma cell frequency in stimulated purified naïve B cells from allo-positive and allo-negative patients in the presence of different doses of QA plus 2.5 μM or 5 μM heme. Data represent means ± standard error of the mean. *P < .05 vs control group; **P < .01 vs control group.
QA increases heme-mediated inhibition of B-cell activation. (A-B) Fold change in plasma cell frequency in 7-day stimulated purified naïve HD B cells in the presence of 2.5 μM QA, AQ, CQ, or DHA without or with 5 μM heme relative to untreated but stimulated cultures (A), or different doses of QA plus 2.5 μM or 5 μM heme (B). (C) Levels of HO-1 expression (MFI) in proliferated B cells of 7-day stimulated B cells treated with different doses of QA without or with 5 μM heme. (D) Fold change in HO-1 expression in stimulated B cells in the presence of 2.5 μM QA, AQ, CQ, or DHA without or with 5 μM free heme relative to untreated but stimulated cultures. (E) Fold change in plasma cell frequency in stimulated purified naïve B cells from allo-positive and allo-negative patients in the presence of different doses of QA plus 2.5 μM or 5 μM heme. Data represent means ± standard error of the mean. *P < .05 vs control group; **P < .01 vs control group.
We next tested the effect of QA on DOCK8 and HO-1 expression in stimulated B cells in the presence of heme. QA had no significant effect on DOCK8 expression (supplemental Figure 4A), but we found a dose-dependent increase in HO-1 expression in proliferated B cells in the presence of heme and QA (fivefold increase at the highest dose; Figure 4C). HO-1 upregulation occurred only in heme plus QA-stimulated B cells, but not with the other heme-binding molecules AQ, CQ, or DHA (Figure 4D). Given that HO-1 inhibits B-cell activation (Figure 3), these data suggest that inhibition of plasma cell differentiation by heme plus QA is likely through induction of HO-1 expression.
Finally, we examined the effect of QA on B cells from allo-negative and allo-positive SCD patients. QA had a weak but significant inhibitory effect on plasma cell differentiation in the absence of heme in both groups of SCD patients (Figure 4E), possibly because of residual heme within SCD B cells. In allo-negative SCD patients, QA further increased heme-mediated suppression of plasma cells (in blue, Figure 4E), but more importantly, it reversed resistance to heme suppression in allo-positive patients (in red), resulting in inhibition of plasma cell differentiation of heme-treated B cells (Figure 4E), thus underscoring its therapeutic potential for decreasing SCD alloimmunization.
Discussion
In this study, we demonstrated that hemolysis regulates human B-cell activation, inhibiting plasmablast and plasma cell differentiation. Mechanistically, heme targeted DOCK8high plasma cells, inhibiting the STAT3 signaling pathway in stimulated B cells. The inhibitory effects of heme on activated B cells were also mediated through HO-1 enzymatic activity and specifically the HO-1 byproducts CO and biliverdin. Compared with nonalloimmunized SCD patients, B cells from alloimmunized SCD patients expressed lower levels of DOCK8 and were less responsive to inhibition by heme and HO-1. These data support our working model in which B-cell intrinsic signals sensing heme and hemolysis control the humoral immune response to allogenic transfusions, and ultimately RBC alloimmunization in SCD patients. This study has thus unraveled a novel mechanism of humoral immunity suppression by hemolysis that has potential for identifying new therapeutic targets as well as B-cell–associated biomarkers of alloimmunization in SCD (Figure 5).
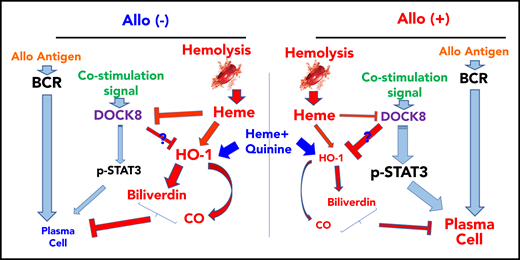
Proposed mechanism of hemolysis-mediated regulation of SCD alloimmunization through inhibition of B-cell differentiation. In allo-negative SCD patients, hemolysis can inhibit B-cell differentiation and subsequent alloimmunization through DOCK8 and HO-1 enzyme activity. In contrast, B cells from allo-positive SCD patients are insensitive to inhibitory effects of hemolysis that results from altered DOCK8 and HO-1 signaling pathways. Heme plus QA can inhibit B-cell activation in both allo-positive and allo-negative patients by targeting HO-1. BCR, B-cell receptor.
Proposed mechanism of hemolysis-mediated regulation of SCD alloimmunization through inhibition of B-cell differentiation. In allo-negative SCD patients, hemolysis can inhibit B-cell differentiation and subsequent alloimmunization through DOCK8 and HO-1 enzyme activity. In contrast, B cells from allo-positive SCD patients are insensitive to inhibitory effects of hemolysis that results from altered DOCK8 and HO-1 signaling pathways. Heme plus QA can inhibit B-cell activation in both allo-positive and allo-negative patients by targeting HO-1. BCR, B-cell receptor.
Several genome-wide association studies have identified genetic variants as potential risk factors for alloimmunization in SCD.8,9,39,40 Most are likely to target steps in the humoral immune response, starting with activation of innate immune antigen-presenting cells through to CD4+ helper T cells and ultimately B cells. Our previous studies have identified abnormal responses in several of these immune subsets in allo-positive SCD patients, including CD16+ monocytes,22 dendritic cells,24 the immunosuppressive regulatory T cells (Tregs)41 and T follicular helper cells.42 Along with this study, these data support a proposed model of a heightened humoral immune response in allo-positive patients that leads to a higher risk of RBC alloimmunization. It is likely that increased alloimmunization risk in allo-positive SCD patients is a result of cumulative impaired heme responses in >1 immune effector cell type, including lower HO-1 expression in CD16+ monocytes leading to inefficient Treg expansion in hemolytic conditions,22 insensitivity to heme-mediated inhibition of dendritic cell maturation,24 and altered B-cell activation in response to heme. No differences were found in hemolysis-associated indices between alloimmunized and nonalloimmunized SCD patients, including total plasma heme, bilirubin, or lactate dehydrogenase levels or reticulocyte percentages, suggesting that it is not hemolysis level per se but rather the immune cell–intrinsic response to hemolysis that is a determinant of alloimmunization risk. RBC destruction and production that results in elevated heme release43-46 occurs mainly in the spleen and/or bone marrow. Thus, B-cell heme response is likely to play a more important role in RBC alloimmunization, which occurs primarily in the spleen rather than in other humoral responses such as vaccine responses, which mostly develop in the lymph nodes or at sites of vaccine delivery (muscle and skin). The differential impact of heme on B-cell development in the various lymphoid organs likely accounts for why no differences are detected in the overall circulating B-cell subset numbers and frequencies between alloimmunized and nonalloimmunized SCD patients.47,48 Differentiation of memory B cells into plasma cells was not affected by heme, although it remains to be determined whether hemolysis affects differentiation of naïve B cells to memory B cells, thus contributing to their low circulating levels in SCD, which have been ascribed to asplenia.49-51
Our finding that heme inhibits human plasma B-cell differentiation in vitro differs from mouse data showing increased plasma B-cell differentiation through Bach2.25 We found very low levels of Bach2 in human B cells before and after stimulation, with no significant change even in the presence of heme (data not shown), suggesting that Bach2 response to heme differs between human and mouse B cells. In the published studies, lipopolysaccharide (LPS) was used to stimulate mouse B cells.25 However, TLR4, the receptor that responds to either LPS or heme,52 is not expressed on human B cells.53,54 Thus, instead of LPS, we used a cocktail of anti-IgG, anti-IgM, anti-CD40, and CpG to activate human B cells. Interestingly, similar B-cell activation cocktails cause mouse B cells to differentiate into plasma cells in vitro. However, addition of heme or heme plus QA did not inhibit mouse plasma cell differentiation (supplemental Figure 5B), and treatment of SCD mice with QA did not lower RBC alloimmunization levels in vivo (supplemental Figure 5C). These data indicate clear differences between human and mouse B-cell response to heme, irrespective of the B-cell activation stimuli.
We have identified an unexpected relationship between DOCK8 expression levels in activated B cells and response to heme. Functionally altered DOCK8 models have been highly informative of the role of DOCK8 in immunity,34,55 as have studies of DOCK8 protein expression levels in immune cells or disease states.56-61 For example, the reduced Treg DOCK8 levels in patients with atopic dermatitis is likely responsible for reduced Treg-derived interleukin-10 (IL-10) and transforming growth factor β expression.57 Low DOCK8 expression can cause impaired neutrophil migration in patients with myelodysplastic syndrome,60 whereas high DOCK8 expression leads to increased leukemic cell survival in acute myeloid leukemia.59 Together with our data, these studies suggest that DOCK8 expression can be a potential biomarker as well as therapeutic target in various disorders.
DOCK8 levels did not correlate with hemolysis levels in SCD patients, consistent with our proposed model that it acts as a heme sensor within B cells. It remains unclear how DOCK8 levels are regulated in B cells. In hepatocellular carcinoma cells, CD147 activation induces DOCK8 expression through SRC signaling and STAT3 phosphorylation.56 Our data indicate that heme inhibits B-cell STAT3 phosphorylation, potentially leading to a decrease in DOCK8high B-cell numbers. Another potential B-cell regulator of DOCK8 expression is miR-34a, shown to inhibit neutrophil DOCK8 expression in myelodysplastic syndrome.60 miR-34a regulates B-cell development by inhibiting the transition of pro-B cells into pre-B cells, and its expression is inhibited by HO-1 enzymatic activity,62 although its effect on naïve B-cell activation and/or whether HO-1 induction by heme can modulate its expression remains unknown.
Our study has uncovered 2 signaling pathways, namely through STAT3 and HO-1, responsible for heme-mediated inhibition of B cells. Whether these pathways interact directly in B cells is not known, although a complex interaction exists in other cell types and disease states. For example, STAT3 in monocytes or macrophages and endothelial cells was shown to mediate HO-1 induction by IL-10 and IL-6,63,64 respectively. STAT3 was also reported to be essential for the protective effects of HO-1 in oxidant-induced lung injury.65-67 The effect of HO-1 on STAT3 activity is more complex. HO-1 inhibited STAT3 activation in endothelial and prostate cancer cells.68-71 In contrast, in various disease models,72-74 HO-1 activated STAT3 by increasing STAT3 phosphorylation. Thus, the interaction between HO-1 and STAT3 likely depends on cell type and disease state. Interestingly, by using a selective SYK inhibitor, which is expected to inhibit the DOCK8/STAT3 signaling pathway,27 we found induction of HO-1 in stimulated B cells (supplemental Figure 6), suggesting that heme may induce HO-1 in B cells by blocking DOCK8 signaling. Future studies are needed to decipher the potential crosstalk between these 2 pathways, specifically in SCD B cells.
Identification of CO and biliverdin, 2 byproducts of HO-1 enzyme activity,23 as potent inhibitors of plasma B-cell differentiation is a key finding of our study and further supports the role of HO-1 enzymatic activity in suppressing B-cell development. The exact mechanism by which CO and biliverdin suppress plasma B-cell differentiation remains unclear. CO mediates its anti-proliferative and anti-inflammatory effects through binding hemoproteins such as soluble guanylate cyclase (sGCS) and P38 MAPK.75 However, the role of either cGMP, produced by sGCS, or the P38 MARK signaling pathway on plasma B-cell differentiation is unclear. In addition to its antioxidant activity, biliverdin can activate aryl hydrocarbon receptor (AhR) signaling.76 AhR is expressed at low levels in resting B cells but is highly upregulated following activation.77 AhR inhibits plasma B-cell differentiation by suppressing several key B-cell transcription factors including Blimp-1, XBP1,78-82 and STAT3 phosphorylation.83 This raises the interesting possibility that the B-cell response to heme in SCD may be through the biliverdin/AhR pathway and that B-cell sensitivity to biliverdin may be an alloimmunization risk factor, opening up the prospect of using biliverdin and/or other AhR agonists as therapeutic candidates for preventing RBC alloimmunization in SCD patients.
An exciting finding of our study is that B-cell response to heme can be modulated by heme-binding small molecules.35-38 Specifically, in the presence of QA, B cells from alloimmunized SCD patients were no longer resistant to the inhibitory effects of heme. This raises the potential for therapeutic novel use of QA for inhibition of alloimmunization in SCD patients. These results also indicate that heme-binding small molecules do not simply neutralize free heme like the heme scavenger hemopexin but rather exhibit potent immune-modulating activity, possibly through forming a complex with heme. In the case of QA, the immunomodulatory mechanism is likely mediated through upregulation HO-1 because heme plus QA induced a fivefold increase in HO-1 levels in stimulated B cells. In this study, we focused on the effects of heme and QA on B-cell differentiation into plasma cells, but whether they impact other B-cell functions such as antigen presentation84,85 or immune regulation47 needs further study. Heme plays a critical role in sickle cell pathophysiology,16-18,21,86-90 and given its immunomodulatory role, altered immune activation may further contribute to sickle complications. Identification of heme-binding small molecules with novel immunomodulatory properties offers the potential for their use for prevention and/or reversion of SCD complications as well as other hemolytic conditions.
For original data, please contact the corresponding authors Hui Zhong at hzhong@nybc.org or Karina Yazdanbakhsh at kyazdanbakhsh@nybc.org.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported, in part, by grants from the National Institutes of Health/National Heart, Lung, and Blood Institute (R01 HL130139, P01 HL149626, and R01 HL145451) (K.Y.), the Hugoton Foundation (H.Z.), a grant from the ASH Bridge Fund (K.Y.), and Bank of New York Mellon (C.A.L. and K.Y.).
Authorship
Contribution: M.P. and W.B. helped conceive the study, performed experiments, and analyzed the data; R.W. performed experiments and analyzed the data; Y.L. helped with the experiments; X.A. and C.A.L. helped with project concept and design; W.B.M., C.M., P.A.S., and D.M. were involved with all aspects of selection, recruitment, and provision of blood samples from patients and controls; K.Y. directed the overall research design and supervised the project; H.Z. conceived and designed the study and interpreted the data; H.Z. and K.Y. wrote the manuscript; and all authors consulted on and contributed to the study.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Hui Zhong, Laboratory of Immune Regulation, New York Blood Center, 310 E 67th St, New York, NY 10065; e-mail: hzhong@nybc.org; and Karina Yazdanbakhsh, Laboratory of Complement Biology, New York Blood Center, 310 E 67th St, New York, NY 10065; e-mail: kyazdanbakhsh@nybc.org.
