

In this issue of Blood, report that leukemia or lymphoma cells with TP53 loss have a selective advantage over time when treated with sublethal doses of a BCL-2 or MCL-1 inhibitor, because of reduced activation of BAX and BAK, but this defect can be overcome with dual treatment with both inhibitors.1
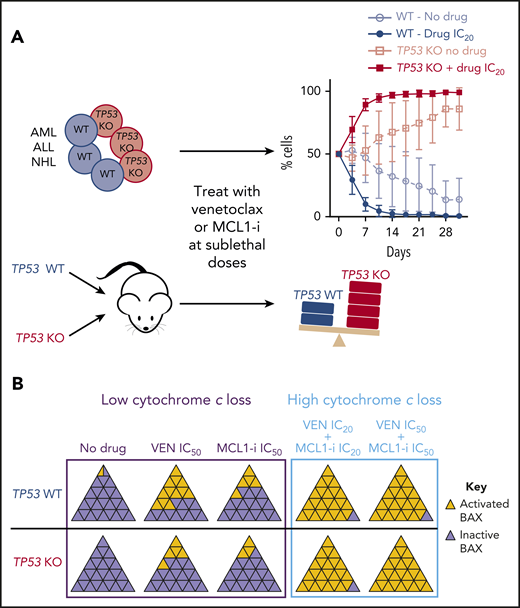
Sublethal dosing of venetoclax or MCL-1 inhibitor selects for TP53 loss clones over time. (A) In AML, ALL, or NHL cell lines in vitro or in AML xenografts, sublethal dosing of either inhibitor leads to progressive selection for the TP53 loss clone over time. (B) TP53 loss is associated with a defect in activating BAX (and BAK) in response to single-agent venetoclax or MCL-1i, but combination therapy causes high levels of activation, with cytochrome c loss leading to apoptosis. WT, wild-type; KO, knockout; MCL-1i, MCL-1 inhibitor; VEN, venetoclax; IC20, 20% inhibitory concentration; IC50, 50% inhibitory concentration. Panel A has been adapted from Figure 2 and panel B is a schematic based on Figure 7 in the article by Thijssen et al that begins on page 2721.
Sublethal dosing of venetoclax or MCL-1 inhibitor selects for TP53 loss clones over time. (A) In AML, ALL, or NHL cell lines in vitro or in AML xenografts, sublethal dosing of either inhibitor leads to progressive selection for the TP53 loss clone over time. (B) TP53 loss is associated with a defect in activating BAX (and BAK) in response to single-agent venetoclax or MCL-1i, but combination therapy causes high levels of activation, with cytochrome c loss leading to apoptosis. WT, wild-type; KO, knockout; MCL-1i, MCL-1 inhibitor; VEN, venetoclax; IC20, 20% inhibitory concentration; IC50, 50% inhibitory concentration. Panel A has been adapted from Figure 2 and panel B is a schematic based on Figure 7 in the article by Thijssen et al that begins on page 2721.
Venetoclax, the first approved drug in the class of BH3-mimetic inhibitors of BCL-2, has emerged as a highly active therapy for both acute myeloid leukemia (AML) and chronic lymphocytic leukemia (CLL). As a BH3-mimetic itself, venetoclax works by binding to BCL-2 and displacing endogenous proapoptotic BH3-mimetics such as BIM, leading to activation of BAX and BAK to induce apoptosis. This activity of venetoclax is downstream of TP53 function, suggesting that venetoclax-induced cell death could remain highly effective, regardless of TP53 loss, consistent with in vitro data in short-term assays in CLL.2 This observation is also consistent with initial clinical observations that venetoclax induces deep remission in CLL, with or without TP53,3 although more recent data show that TP53 loss leads to a shorter duration of response.4 In AML also, despite the initial response, TP53 mutations have been associated with early acquired resistance, often accompanied by increased allele frequency of the mutation or selection for biallelic loss.5 These findings suggest that short-term killing with venetoclax may not be substantially affected by TP53 loss, but that, over time, the TP53 mutant clones are likely to have an advantage.
The work of Thijssen et al sheds further light on this conundrum. Starting from the clinical observations that TP53-mutant AML can have a deep response to venetoclax, yet expansion of TP53-mutant clones occurred between baseline and day 8 in 4 nonresponding patients, Thijssen et al deleted TP53 in AML cell lines to explore the impact on venetoclax sensitivity. They found that TP53 deletion had relatively little effect in short-term culture, but marked outgrowth of the TP53-aberrant clone occurred in longer term culture under selection with continuous sublethal venetoclax concentrations (see figure panel A). Sublethal venetoclax similarly favored outgrowth of TP53 mutant clones in engrafted mice, as well as in acute lymphoblastic leukemia (ALL) and non-Hodgkin lymphoma (NHL) cell lines. This outgrowth was not associated with acquired venetoclax resistance, with venetoclax-induced DNA damage that may require upstream TP53 function, or with altered baseline or venetoclax-induced mitochondrial function. Instead, Thijssen et al found that TP53 deficiency reduced activation of BAX and BAK in response to venetoclax, despite unchanged protein levels, and led to early resistance to apoptosis induction (see figure panel B).
They were then interested in whether this mechanism of resistance applies to other BH3-mimetic inhibitors, specifically an MCL-1 inhibitor. Consistent with a role for TP53, the 2 top hits in their CRISPR/Cas9 screening for MCL-1 inhibitor resistance were TP53 and BAX, both of which were also identified in a similar earlier screening for mediators of venetoclax resistance.6 They then performed similar experiments as above with the MCL-1 inhibitor and obtained similar results (see figure panel A), suggesting that this effect of TP53 loss could be a class effect of BH3-mimetic inhibitors.
But why? Previous work has shown that TP53 increases expression of the endogenous proapoptotic BH3 proteins NOXA, PUMA, and BIM. Loss of TP53 leading to decreased expression of these proteins could therefore reduce their release from BCL-2 or MCL-1 in response to the BH3-mimetic drugs, leading to reduced BAX/BAK activation, just as Thijssen et al observed (see figure panel B). In support of this theory, they show a similar phenotype in cells with triple knockout of NOXA, PUMA, and BIM.
What then are the clinical implications? A first question is whether venetoclax levels achieved in patients are sublethal and therefore are likely to select TP53-aberrant clones. Thijssen et al chose drug concentrations to span the clinically achieved venetoclax concentrations, and, given the variability likely in patients and the degree to which the clinical observations match their findings, it seems likely that in vivo drug levels are sublethal at least some of the time, raising the question of whether studies of increased venetoclax doses are warranted. In the phase 1 study of CLL, progression-free survival trended better at doses of >400 mg.7 A second question is whether these findings apply more broadly than only to AML. Thijssen et al looked at ALL and aggressive NHL cell lines, where the selection for the TP53 loss clones appears somewhat less than in AML, possibly because of slower proliferation. If so, selection in CLL may be even less, although the clinical observations in CLL to date align with the findings, albeit over a longer time frame. Data on clonal evolution of TP53 at CLL relapse after venetoclax remain sparse and further analyses will be informative.
The third and most important question centers on how we overcome this selection of TP53 loss clones. The deficiency in BAX/BAK activation was completely abrogated by combining venetoclax with the MCL-1 inhibitor (see figure panel B), but not by adding cytarabine or decitabine. Whether other targeted therapies in combination with venetoclax would overcome this early apoptotic defect in BAX/BAK activation remains to be determined. For example, in CLL, the combination of venetoclax with BTK inhibitors is under intensive study, yet it is not obvious that a BTK inhibitor would overcome the apoptotic defect associated with TP53 loss. It is likely that each proposed combination will have to be specifically characterized.
These data add to the increasing body of literature supporting the potential of combination BCL-2 and MCL-1 inhibitors in the clinic, certainly in AML,8,9 but potentially also in CLL.10 Multiple MCL-1 inhibitors are currently in the clinic, but caution is warranted, given that the phase 1 trial of AMG397 was put on hold because of cardiac side effects. The development strategy may therefore be complex, but the data reported by Thijssen et al suggest that lower doses in combination may maintain efficacy with potentially lower toxicity.
The particular excitement raised by the data from Thijssen et al lies in how well they explain the emerging clinical observations in patients with TP53-aberrant disease. Although much work remains to improve the prognosis for these patients, this research provides a model system for testing combinations and further rationale for the pairing of venetoclax with MCL-1 inhibitors in AML and CLL.
Conflict-of-interest disclosure: J.R.B. has served as a consultant for Abbvie, Acerta/Astra-Zeneca, Beigene, Bristol-Myers Squibb/Juno/Celgene, Catapult, Genentech/Roche, Eli Lilly, Janssen, MEI Pharma, Morphosys AG, Nextcea, Novartis, Pfizer, and Rigel.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal