

Key Points
Bimodal Bfl-1 expression in lymphomas mediates resistance to Mcl-1 and Bcl-2 inhibitors.
Bfl-1 is rapidly suppressed upon transient CDK9 inhibition, providing a clinical opportunity for CDK9 inhibitors in Bfl-1+ lymphomas.

Abstract
BH3 mimetics like venetoclax target prosurvival Bcl-2 family proteins and are important therapeutics in the treatment of hematological malignancies. We demonstrate that endogenous Bfl-1 expression can render preclinical lymphoma tumor models insensitive to Mcl-1 and Bcl-2 inhibitors. However, suppression of Bfl-1 alone was insufficient to fully induce apoptosis in Bfl-1–expressing lymphomas, highlighting the need for targeting additional prosurvival proteins in this context. Importantly, we demonstrated that cyclin-dependent kinase 9 (CDK9) inhibitors rapidly downregulate both Bfl-1 and Mcl-1, inducing apoptosis in BH3-mimetic–resistant lymphoma cell lines in vitro and driving in vivo tumor regressions in diffuse large B-cell lymphoma patient-derived xenograft models expressing Bfl-1. These data underscore the need to clinically develop CDK9 inhibitors, like AZD4573, for the treatment of lymphomas using Bfl-1 as a selection biomarker.
Introduction
Hematological cancers notoriously exploit the mitochondrial apoptotic machinery for survival by overexpressing antiapoptotic Bcl-2 family proteins.1 This survival benefit also confers a therapeutic vulnerability whereby targeting prosurvival proteins with BH3 mimetics induces apoptosis and tumor cell death. The selective Bcl-2 inhibitor venetoclax (Venclexta) is approved for treating different forms of leukemia,2,3 and emerging BH3 mimetics selectively targeting Mcl-1 are also in clinical development for the treatment of hematological malignancies.4
The extent and duration of antitumor responses to selective BH3 mimetics may be limited by compensatory adaptations inherent in the overlapping affinities of the Bcl-2 family antiapoptotic proteins for multiple prodeath BH3-only proteins.5 Previous reports have shown that resistance to continuous navitoclax exposure in preclinical B-cell lymphoma models is mediated through increased levels of Mcl-1, Bcl-xL, and Bfl-1 antiapoptotic proteins that redundantly sequester the proapoptotic protein Bim.6,7 A similar clinical observation was made in a venetoclax-relapsed chronic lymphocytic leukemia (CLL) patient who presented a dominant Bcl-xL–overexpressing subclone.8 Although the arsenal of BH3 mimetics in clinical development will hopefully combat some adaptations to venetoclax, to our knowledge none have activity against Bfl-1.1
Given the difficulty in developing direct small molecule inhibitors of Mcl-1 until recently,9-12 orthogonal approaches to indirectly modulate this high-value oncology target have been explored.13 One clinically validated approach is the acute inhibition of cyclin-dependent kinase 9 (CDK9).14-16 The resulting transient suppression of transcriptional elongation via reduced activity of RNA polymerase II (RNAP2) triggers rapid Mcl-1 depletion due to its short-lived messenger RNA (mRNA) and protein. Similar to Mcl-1, other labile oncogenes are suppressed from CDK9 inhibition, including MYC.17 In Mcl-1–dependent cells, CDK9 inhibition causes a classical apoptogenic response, characterized by mitochondrial outer membrane permeabilization (MOMP), caspase activation, and eventual cell death. The strong correlation in preclinical activity for the selective Mcl-1 and CDK9 inhibitors, AZD5991 and AZD4573, respectively, highlights Mcl-1 suppression as the predominant mechanism driving CDK9-inhibitor antitumor responses upon acute exposure.16 Despite this compelling evidence, a subset of cancer cell lines was highly sensitive to CDK9 but not Mcl-1 inhibition, suggesting that CDK9 may regulate additional intrinsic apoptotic regulators beyond Mcl-1. Although levels of most other Bcl-2 family proteins are relatively static following acute CDK9 inhibition,16 Bfl-1 is reported to have a short protein half-life comparable to Mcl-1.18-20 To this extent, we hypothesized that CDK9 inhibition rapidly modulated Bfl-1 to trigger apoptosis in Bfl-1–dependent cancers that were insensitive to Mcl-1 inhibition alone.
Materials and methods
Western blot analysis
Cell pellets containing ∼5 × 106 viable cells were collected. Whole-cell lysates were prepared by addition of 0.1 mL of NP-40 lysis buffer (Thermo Fisher Scientific) with addition of protease and phosphatase inhibitors (Sigma-Aldrich). Protein concentration was measured using a BCA protein assay kit (Thermo Fisher Scientific) and lysates were equilibrated to 2 mg/mL containing 1× NuPAGE LDS sample buffer (Thermo Fisher Scientific) and 1× NuPAGE reducing reagent (Thermo Fisher Scientific). Samples were heated at 95°C for 6 minutes prior to loading ∼40 µg of lysate onto NuPAGE 4% to 12% 1.5-mm Bis-Tris gels (Thermo Fisher Scientific). Sodium dodecyl sulfate–polyacrylamide gel electrophoresis was run at 110 V for 2 hours prior to gel transfer onto 0.45-µm nitrocellulose membranes (Thermo Fisher Scientific) via wet transfer using NuPAGE transfer buffer (Thermo Fisher Scientific) containing 10% methanol for 3 hours at 30 V. Membranes were blocked in either 5% nonfat milk or 5% bovine serum albumin for 1 hour prior to primary antibody addition overnight at 4°C. Antibodies used for experimental analysis are listed in supplemental Table 2 (available on the Blood Web site). Secondary antibodies were added at 1/5000 dilution for 1 hour prior to incubation with Immobilon Western Chemiluminescent HRP Substrate (EMD Millipore). Images were captured using a FugiFilm ImageQuant CCD camera (GE Healthcare). Densitometric quantification was performed using 1-dimensional gel analysis in ImageQuant TL software (GE Healthcare) with rolling ball background subtraction. Protein expression across cell-line panels was performed by normalizing to TMD8 and Karpas-422 control cell lysates on every gel. Relative expression of Bfl-1, Mcl-1, Bcl-2, and Bcl-xL levels in lymphoma cell lines was normalized to TMD8 lysate as 100%. Relative expression of Bcl-w levels in lymphoma cell lines was normalized to Karpas-422 lysate as 100%.
Caspase activation and cell-viability assays
Lymphoma cell lines were seeded overnight on white opaque plates (384w; Corning) at 5000 cells per well in a 30-µL volume. Compounds were dosed using 8-point half-log dilutions with a 3-µM top concentration using an Echo 555 acoustic liquid dispenser (Labcyte). Combinations were dosed using 5-point half-log dilutions of AZD5991 with a 0.33-µM top concentration plus a fixed 0.33-µM dose of venetoclax. For measurement of caspase activation, cells were incubated with compounds for 6 hours followed by addition of Caspase-Glo 3/7 (Promega) following the vendor-supplied protocol. The percentage of caspase activation was calculated by normalizing drug-induced luminescent measurements to maximum (100% mixture of 0.5 µM AZD5991 and 0.5 µM AZD4320 inhibitors) and minimum (dimethyl sulfoxide [DMSO] only) controls. Cell viability was measured 24 hours after drug incubation using CellTiter-Glo (Promega). The percentage of viability was calculated by normalizing drug-induced luminescent measurements to a negative control (DMSO only). Dose-response curves were fitted using nonlinear regression least-squares fit of [inhibitor] vs response in GeneData Screener or GraphPad Prism.
In vivo studies
Female CB17 SCID mice were purchased from Charles River Laboratories for xenograft studies conducted at AstraZeneca in accordance with animal research guidelines from the National Institutes of Health and the AstraZeneca Institutional Animal Care and Use Committee. A total of 5 × 106 OCILY10 and TMD8 cells, respectively, were injected subcutaneously in the right flank of mice, with initiation of drug treatments upon tumor volumes reaching ∼200 mm3. Diffuse large B-cell lymphoma (DLBCL) patient-derived xenograft (PDX) efficacy studies were performed at CrownBio in NOD-SCID mice. For all animal studies, AZD4573 was formulated in 2%/30%/68% 131 mix of N,N-dimethylacetamide, PEG-400, and 1% (v/v) Tween 80 and dosed at 15 mg/kg, twice daily with a 2-hour split dose, 2 days on/5 days off. AZD5991 was formulated in 30% 2-hydroxylpropyl-β-cyclodextrin at pH 9 and dosed IV at 60 mg/kg once per week.
For pharmacodynamic analysis, subcutaneous tumors were carefully harvested from euthanized animals and dissociated using an Omni Prep Multi-Sample Homogenizer (Omni, Inc). A total of 0.1 mL of NP-40 lysis buffer was added for each 10 mg of tumor tissue. Lysate was extracted from homogenized tissue via centrifugation at 14 000 RPM for 0.25 hours at 4°C, transferring 0.2 mL of material from the nonlipid fraction. Protein expression assays were performed as described n the "Western blot analysis" section.
Statistical analysis
All statistical analysis was performed via the unpaired Student t test in GraphPad Prism.
Results
Bfl-1 is overexpressed in lymphomas and facilitates de novo resistance to BH3 mimetics
The cancer cell-line encyclopedia (CCLE) and internal western blot data were used to evaluate the prevalence of BCL2A1/Bfl-1 mRNA and protein levels across preclinical hematological models. BCL2A1 expression was elevated in lymphoma compared with myeloid and leukemia lineages (Figure 1A) although Bfl-1 protein was detected in only a subset of lymphoma cell lines (17 of 52, “Bfl-1+ lymphomas”), 41% of which were classified as the activated B-cell DLBCL (ABC-DLBCL) (Figure 1B; supplemental Figure 1A; supplemental Table 1). Only 1 of 30 nonlymphoid blood cancer cell lines expressed detectable Bfl-1 protein by western blot (supplemental Figure 1B). Given the limited availability of validated Bfl-1 antibodies,21 we confirmed the western blot results with an orthogonal mesoscale discovery (MSD) immunoassay measuring Bfl-1 protein levels under nondenaturing conditions (supplemental Figure 1C). Interestingly, both of the mRNA and protein levels exhibited a bimodal distribution pattern for Bfl-1 across lymphoma tumor models in contrast with the more uniform distribution of other antiapoptotic Bcl-2 family members (Figure 1B-D). These results also aligned with the Genome-Tissue Expression (GTEx) portal that showed minimal BCL2A1 expression in nonhematopoietic primary compartments similar to BCL2 but distinct from MCL1, BCL2L1 (Bcl-xL), and BCL2L2 (Bcl-w) (supplemental Figure 1D). Increased Bfl-1 expression has been reported as a mechanism of resistance to cancer therapies, although its prevalence in mediating intrinsic resistance is lesser studied, particularly in the context of lymphoma.6,7,22,23 Thus, we investigated the relationship between Bfl-1 expression and the sensitivity of BH3 mimetics across a lymphoma cell-line panel. Relative to lymphoma cell lines without detectable Bfl-1 protein (“Bfl-1− lymphomas”), Bfl-1+ lymphomas exhibited reduced sensitivity, as measured by cleaved caspase-3/7 activation (“caspase activation”), to BH3 mimetics targeting Mcl-1 (AZD5991) and Bcl-2 (venetoclax) (Figure 1E), which translated to significantly reduced cell death compared with that achieved in Bfl-1− lymphoma cell lines (Figure 1F). The sensitivity of a selective BH3 mimetic targeting Bcl-xL (referred to as “compound 15”) was slightly reduced in Bfl-1+ lymphomas, although only 12 of 52 cell lines were intrinsically sensitive to short-term Bcl-xL inhibition (50% effective concentration [EC50] <0.5 µM) irrespective of Bfl-1 status (supplemental Figure 1E). Furthermore, even the combination of Mcl-1 and Bcl-2 inhibition, which has previously been shown to overcome monotherapy BH3-mimetic resistance,9,10,24 exhibited lower caspase activation in Bfl-1+ lymphomas, as determined by both increased EC50 and reduced area under curve (AUC) parameters (Figure 1G-H). Collectively, these results demonstrate that Bfl-1 expression can mediate resistance to Bcl-2, Mcl-1, and Bcl-xL inhibitors.
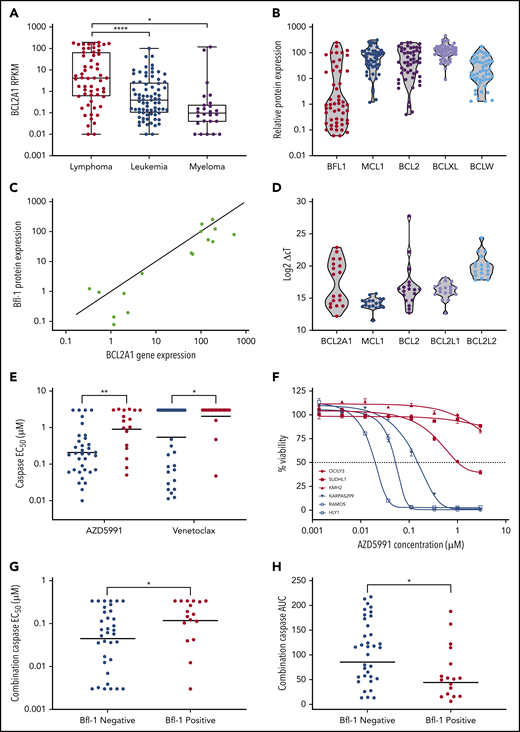
Bfl-1 is overexpressed in lymphomas and confers resistance to BH3 mimetics. (A) Reads per kilobase of transcript per million mapped reads (RPKM) for BCL2A1 across hematological cancer cell lines from the CCLE RNA-sequencing data set. (B) Violin plot for relative Bcl-2 family antiapoptotic protein levels across 52 lymphoma cell lines. Data were acquired from densitometric measurements of western blot band intensities from supplemental Figure 1A. (C) Correlation between BCL2A1 mRNA and Bfl-1 protein levels in a subset of 18 evaluated lymphoma cell lines. BCL2A1/Bfl-1 levels were normalized relative to the TMD8 cell line as 100%. Solid line indicates y = x-axis unity. (D) BCL2A1, MCL1, BCL2, BCL2L1, BCL2L2 mRNA abundance in lymphoma cell lines. (E) AZD5991 and venetoclax were assessed for caspase activation after 6 hours of drug incubation across the lymphoma cell line panel. Each dot represents individual cell lines that are either Bfl-1+ (red) or Bfl-1− (blue). Solid line indicates the geometric mean 50% effective concentration (EC50). (F) Cell viability of Bfl-1− (blue) or Bfl-1+ (red) cell lines in response to a dose response of AZD5991 treated for 24 hours. (G-H) Lymphoma cell lines were treated with a fixed 0.33-µM dose of venetoclax combined with a dose response of AZD5991 for 6 hours before caspase activation was assessed. Each dot represents the caspase EC50 (G) or area under the curve (AUC) parameters (H) of individual cell lines that are Bfl-1+ (red) or Bfl-1− (blue). Solid line indicates the geometric mean EC50 (G) or AUC (H). *P < .05; **P < .01; ****P < .0001.
Bfl-1 is overexpressed in lymphomas and confers resistance to BH3 mimetics. (A) Reads per kilobase of transcript per million mapped reads (RPKM) for BCL2A1 across hematological cancer cell lines from the CCLE RNA-sequencing data set. (B) Violin plot for relative Bcl-2 family antiapoptotic protein levels across 52 lymphoma cell lines. Data were acquired from densitometric measurements of western blot band intensities from supplemental Figure 1A. (C) Correlation between BCL2A1 mRNA and Bfl-1 protein levels in a subset of 18 evaluated lymphoma cell lines. BCL2A1/Bfl-1 levels were normalized relative to the TMD8 cell line as 100%. Solid line indicates y = x-axis unity. (D) BCL2A1, MCL1, BCL2, BCL2L1, BCL2L2 mRNA abundance in lymphoma cell lines. (E) AZD5991 and venetoclax were assessed for caspase activation after 6 hours of drug incubation across the lymphoma cell line panel. Each dot represents individual cell lines that are either Bfl-1+ (red) or Bfl-1− (blue). Solid line indicates the geometric mean 50% effective concentration (EC50). (F) Cell viability of Bfl-1− (blue) or Bfl-1+ (red) cell lines in response to a dose response of AZD5991 treated for 24 hours. (G-H) Lymphoma cell lines were treated with a fixed 0.33-µM dose of venetoclax combined with a dose response of AZD5991 for 6 hours before caspase activation was assessed. Each dot represents the caspase EC50 (G) or area under the curve (AUC) parameters (H) of individual cell lines that are Bfl-1+ (red) or Bfl-1− (blue). Solid line indicates the geometric mean EC50 (G) or AUC (H). *P < .05; **P < .01; ****P < .0001.
Bfl-1+ lymphomas are highly codependent on Mcl-1 for optimal survival
Given the reduction in apoptogenic potential of Mcl-1 or Bcl-2 inhibition in Bfl-1+ lymphomas, we used RNA interference (RNAi) methods to determine the dependency of lymphoma tumor models on Bfl-1 expression for survival. RNAi-mediated silencing of BCL2A1 in the anaplastic large cell lymphoma (ALCL) cell line SUDHL1 caused a dose-dependent increase in apoptotic cells, quantified by MOMP (percentage of TMRE− cells), whereas monotherapy or combination treatment of Mcl-1 and Bcl-2 inhibitors had no effect. (Figure 2A). However, the magnitude of MOMP resulting from near-complete Bfl-1 protein knockdown was <60%, which was comparable to that from Bfl-1 knockdown in 3 models of ABC-DLBCL, suggesting that many Bfl-1+ lymphomas may be codependent on other Bcl-2 family prosurvival proteins to evade complete cell death (supplemental Figure 2A-B).
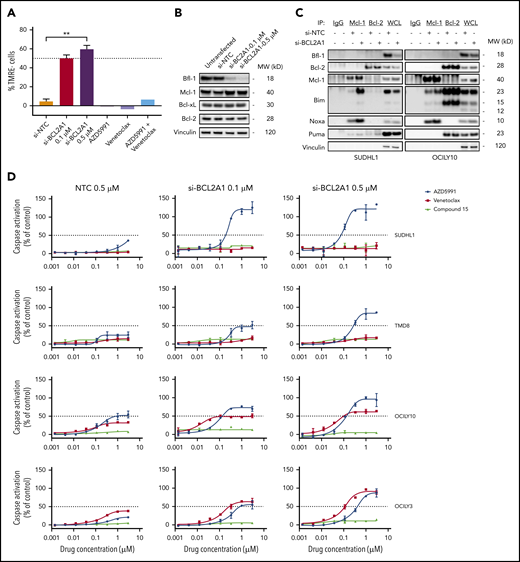
Bfl-1+ lymphomas are codependent on additional antiapoptotic Bcl-2 family proteins for survival. (A) SUDHL1 cells were transfected with nontargeting control (NTC) or BCL2A1 siRNA for 72 hours, or treated with 0.5 µM AZD5991, 0.5 µM venetoclax, or 0.5 µM AZD5991 plus 0.5 µM venetoclax for 8 hours followed by flow cytometric measurement of MOMP. Results are plotted as TMRE− cells, normalized to untreated cells (0%). (B) Western blot analysis for Bfl-1 protein knockdown efficiency and effects on Bcl-2 family prosurvival proteins following 72 hours of BCL2A1 transfection in SUDHL1 cells. Loading control, vinculin. (C) Mcl-1 and Bcl-2 were immunoprecipitated from whole-cell lysates (WCL) in SUDHL1 and OCILY10 cell lines following 72 hours transfection with 0.5 µM nontargeting control or 0.5 µM BCL2A1 siRNA and immunoblotted for the indicated Bcl-2 family proteins. Species specific isotype controls are included in lanes 1 (mouse IgG) and 2 (rabbit IgG). (D) Lymphoma cell lines were transfected with the indicated concentrations of NTC or BCL2A1 siRNA for 72 hours followed by treatment of 6 hours with a dose titration of Mcl-1 (AZD5991), Bcl-2 (venetoclax) or Bcl-xL (compound 15) inhibitors before measuring caspase activation. **P < .01.
Bfl-1+ lymphomas are codependent on additional antiapoptotic Bcl-2 family proteins for survival. (A) SUDHL1 cells were transfected with nontargeting control (NTC) or BCL2A1 siRNA for 72 hours, or treated with 0.5 µM AZD5991, 0.5 µM venetoclax, or 0.5 µM AZD5991 plus 0.5 µM venetoclax for 8 hours followed by flow cytometric measurement of MOMP. Results are plotted as TMRE− cells, normalized to untreated cells (0%). (B) Western blot analysis for Bfl-1 protein knockdown efficiency and effects on Bcl-2 family prosurvival proteins following 72 hours of BCL2A1 transfection in SUDHL1 cells. Loading control, vinculin. (C) Mcl-1 and Bcl-2 were immunoprecipitated from whole-cell lysates (WCL) in SUDHL1 and OCILY10 cell lines following 72 hours transfection with 0.5 µM nontargeting control or 0.5 µM BCL2A1 siRNA and immunoblotted for the indicated Bcl-2 family proteins. Species specific isotype controls are included in lanes 1 (mouse IgG) and 2 (rabbit IgG). (D) Lymphoma cell lines were transfected with the indicated concentrations of NTC or BCL2A1 siRNA for 72 hours followed by treatment of 6 hours with a dose titration of Mcl-1 (AZD5991), Bcl-2 (venetoclax) or Bcl-xL (compound 15) inhibitors before measuring caspase activation. **P < .01.
Upon Bfl-1 knockdown in SUDHL1 cells, we noticed slightly elevated Mcl-1 protein levels with no observable changes in Bcl-2 or Bcl-xL abundance (Figure 2B). Elevated Mcl-1 protein levels can result from stabilization due to increased sequestration of BH3-only proteins,25 so we therefore compared the levels of Mcl-1 and Bcl-2 in protein complex with the activator BH3 only protein, Bim, upon Bfl-1 knockdown. The levels of Bim in complex with Mcl-1 increased across the 4 Bfl-1+ lymphoma cell lines tested whereas levels of Bim in complex with Bcl-2 only marginally increased in 2 of 4 cell lines (supplemental Figure 2C). Furthermore, levels of Noxa in complex with Mcl-1 were also enriched, whereas other BH3-only proteins with affinity for Bfl-1, like Puma, remained mostly static following Bfl-1 depletion, highlighting a codependence on Bfl-1+ lymphomas favoring Mcl-1 (Figure 2C). Additionally, we also interrogated the phenotype of Mcl-1, Bcl-2, or Bcl-xL pharmacological inhibition in combination with dose-dependent Bfl-1 knockdown. In each model, AZD5991 treatment demonstrated an enhanced apoptogenic response that coincided with reductions in Bfl-1 protein levels from small-interfering RNA (siRNA) treatment. On the other hand, the reduction in Bfl-1 levels did not sensitize these lymphoma cell lines to a selective Bcl-xL inhibitor and only marginally enhanced the response to venetoclax, particularly in the OCILY3 model that harbors a BCL2 amplification (Figure 2D). Collectively, these results suggest that inhibiting both Bfl-1 and Mcl-1 is necessary and sufficient to maximize the antitumor response in Bfl-1–expressing lymphomas.
Acute CDK9 inhibition induces intrinsic apoptosis in Bfl-1+ lymphomas
Previous reports have demonstrated a positive correlation in antitumor pharmacological responses between CDK9 and Mcl-1 inhibitors across diverse cancer cell-line panels.16 Thus, we interrogated whether CDK9 inhibitors likewise would be similarly ineffective against Bfl-1+ lymphomas. Across a lymphoma cell-line panel, the CDK9 inhibitor AZD4573 displayed a strong concordance in apoptotic sensitivity with AZD5991 in Bfl-1− lymphomas (caspase EC50, R2 = 0.72) (Figure 3A). Conversely, the correlation was substantially weaker in Bfl-1+ lymphomas as >75% of cell lines were >10-fold more sensitive to AZD4573 (R2 = 0.21) (Figure 3A-B; supplemental Table 1). Notably, only 2 Bfl-1+ lymphomas were resistant to AZD4573 (EC50 of >3 µM) although the level of Bcl-xL expression in each model may have blunted an apoptotic response (supplemental Table 1). CDK9 inhibition with AZD4573 induced near-complete MOMP across multiple Bfl-1+ lymphomas of mixed lineages, including DLBCL, ALCL, and Hodgkin lymphoma, whereas AZD5991 derived only a partial apoptotic response in the same models (Figure 3C). Similar results were obtained using other clinical-stage CDK9 and Mcl-1 inhibitors (dinaciclib14 and S63845,12 respectively) (supplemental Figure 3A). Additionally, we ectopically expressed BCL2A1 in the Mcl-1–dependent acute myeloid leukemia (AML) cell line MV411, which dramatically reduced the magnitude of caspase activation from AZD5991 but not AZD4573 treatment (Figure 3D-E). Collectively, these results indicate that the differential response between CDK9 and Mcl-1 inhibitors in certain preclinical lymphoma models is class-specific and involves Bfl-1 expression.
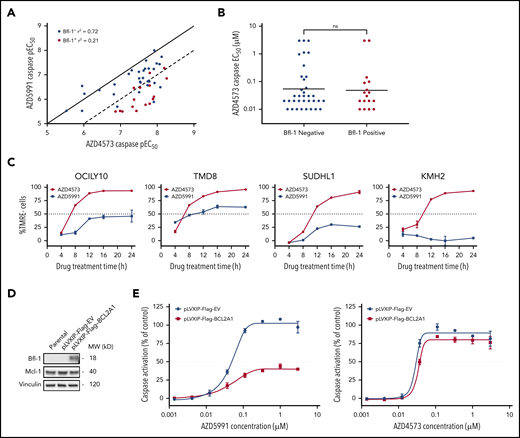
CDK9 inhibitors are highly effective at inducing apoptosis in Bfl-1+ lymphomas. (A) AZD4573 and AZD5991 6-hour caspase negative logarithmic 50% effective concentration (pEC50) correlation across 52 lymphoma cell lines. Dots represent individual cell lines, colored by Bfl-1− (blue) or Bfl-1+ (red) protein expression as determined from western blot analysis. Solid and dashed lines illustrate y = x (1:1) and y = x−1 (1:10) axis unity, respectively. (B) Apoptotic sensitivity of lymphoma cell lines to AZD4573 after 6-hour treatment assessed by caspase EC50 determination. (C) Kinetics of MOMP in Bfl-1+ lymphomas over time following incubation with 0.1 µM AZD4573 or 0.5 µM AZD5991, respectively. (D) Western blot analysis for Bfl-1 and Mcl-1 protein expression in MV411 parental, pLVXIP-Flag-empty vector or pLVXIP-Flag-BCL2A1 cell lines. Loading control, vinculin. (E) AZD5991 (left graph) and AZD4573 (right graph) were treated at the indicated concentrations for 6 hours followed by caspase-3/7 measurement in MV411 pLVXIP-Flag-EV or pLVXIP-Flag-BCL2A1 stable cell lines. ns, no significance.
CDK9 inhibitors are highly effective at inducing apoptosis in Bfl-1+ lymphomas. (A) AZD4573 and AZD5991 6-hour caspase negative logarithmic 50% effective concentration (pEC50) correlation across 52 lymphoma cell lines. Dots represent individual cell lines, colored by Bfl-1− (blue) or Bfl-1+ (red) protein expression as determined from western blot analysis. Solid and dashed lines illustrate y = x (1:1) and y = x−1 (1:10) axis unity, respectively. (B) Apoptotic sensitivity of lymphoma cell lines to AZD4573 after 6-hour treatment assessed by caspase EC50 determination. (C) Kinetics of MOMP in Bfl-1+ lymphomas over time following incubation with 0.1 µM AZD4573 or 0.5 µM AZD5991, respectively. (D) Western blot analysis for Bfl-1 and Mcl-1 protein expression in MV411 parental, pLVXIP-Flag-empty vector or pLVXIP-Flag-BCL2A1 cell lines. Loading control, vinculin. (E) AZD5991 (left graph) and AZD4573 (right graph) were treated at the indicated concentrations for 6 hours followed by caspase-3/7 measurement in MV411 pLVXIP-Flag-EV or pLVXIP-Flag-BCL2A1 stable cell lines. ns, no significance.
Acute exposure to CDK9 inhibitors selectively modulates labile antiapoptotic proteins Bfl-1 and Mcl-1
Bfl-1 is reported to have a short half-life due to highly coordinated proteasomal regulation, like Mcl-1,17-19 which prompted us to evaluate whether CDK9 inhibitors were indirectly and rapidly downmodulating Bfl-1 in addition to Mcl-1. Transient CDK9 inhibition with AZD4573 in OCILY10 cells triggered a dose-dependent suppression of both Bfl-1 and Mcl-1 proteins, with limited to no effect on Bcl-2 or Bcl-xL levels, in DLBCL, ALCL, and Hodgkin lymphoma cell lines (Figure 4A; supplemental Figure 4A-B). Additional CDK9 inhibitors dinaciclib and AZ’557626 exhibited similar effects as AZD4573 (Figure 4B), whereas CDK4/6 inhibition with palbociclib revealed no changes in Bcl-2 family antiapoptotic protein levels (supplemental Figure 4C), suggesting that Bfl-1 modulation is a class-specific effect from inhibiting CDK9. The reduction in Bfl-1 levels was also quantitively measured using a Bfl-1 MSD immunoassay, and in this context Bfl-1 protein levels were reduced approximately 90% following treatment with AZD4573 for 6 hours (Figure 4C). Importantly, AZD4573 effects on Bfl-1 aligned with equipotent reductions in phosphorylation of the direct substrate of CDK9, serine 2 in the C-terminal domain of RNA polymerase 2 (pSer2-RNAP2), while showing no inhibitory effect on putative CDK127 and CDK4/628 biomarkers pPP1a T320 and pRB S807-811 (Figure 4A), respectively, underscoring that selective inhibition of CDK9 mediates the reduction in Bfl-1.
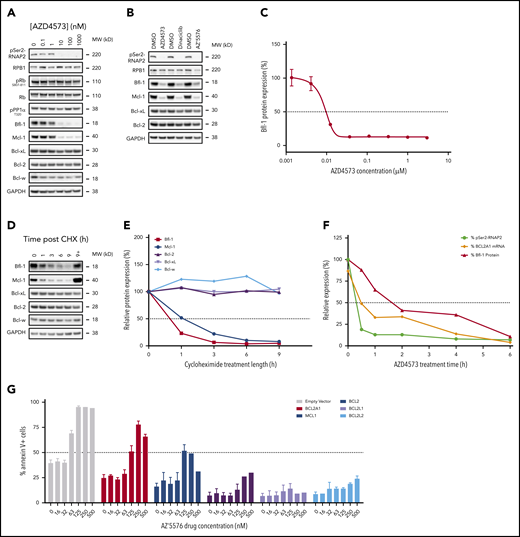
Acute CDK9 inhibition depletes labile antiapoptotic proteins Bfl-1 and Mcl-1. (A) Western blot analysis of CDK9, CDK1, and CDK4/6 putative biomarkers and Bcl-2 family proteins in OCILY10 cells treated with the indicated doses of AZD4573 for 6 hours. Loading control, glyceraldehyde-3-phosphate dehydrogenase (GAPDH). (B) OCILY10 cells were treated with 0.1 µM AZD4573, 0.1 µM dinaciclib, or 1 µM AZ’5576 for 6 and pSer2-RNAP2 and antiapoptotic biomarker expression was assessed by western blot. Loading control, GAPDH. (C) Measurement of Bfl-1 protein levels using MSD immunodetection following 6 hours of AZD4573 exposure in OCILY10 cells. Expression is normalized to DMSO-only treated cells (100%). (D) Western blot analysis of antiapoptotic Bcl-2 family protein levels in OCILY10 cells following exposure to 10 µg/mL cycloheximide (CHX) or CHX plus 10 µM MG132 for the indicated durations. MG132 treatment is indicated by “+”. Loading control, GAPDH. (E) Densiometric quantification of panel D, expression is relative to untreated (time 0 hours, 100%) cells. (F) Measurement of pSer2-RNAP2, BCL2A1 mRNA and Bfl-1 protein levels in OCILY10 cells at the indicated time points following 0.1 µM AZD4573 treatment. (G) Eµ-Myc lymphoma cells ectopically expressing BCL2A1, MCL1, BCL2, BCL2L1, or BCL2L2 prosurvival genes were treated with the CDK9 inhibitor AZ’5576 at the indicated concentrations for 24 hours. Apoptosis was measured by flow cytometric annexin V staining.
Acute CDK9 inhibition depletes labile antiapoptotic proteins Bfl-1 and Mcl-1. (A) Western blot analysis of CDK9, CDK1, and CDK4/6 putative biomarkers and Bcl-2 family proteins in OCILY10 cells treated with the indicated doses of AZD4573 for 6 hours. Loading control, glyceraldehyde-3-phosphate dehydrogenase (GAPDH). (B) OCILY10 cells were treated with 0.1 µM AZD4573, 0.1 µM dinaciclib, or 1 µM AZ’5576 for 6 and pSer2-RNAP2 and antiapoptotic biomarker expression was assessed by western blot. Loading control, GAPDH. (C) Measurement of Bfl-1 protein levels using MSD immunodetection following 6 hours of AZD4573 exposure in OCILY10 cells. Expression is normalized to DMSO-only treated cells (100%). (D) Western blot analysis of antiapoptotic Bcl-2 family protein levels in OCILY10 cells following exposure to 10 µg/mL cycloheximide (CHX) or CHX plus 10 µM MG132 for the indicated durations. MG132 treatment is indicated by “+”. Loading control, GAPDH. (E) Densiometric quantification of panel D, expression is relative to untreated (time 0 hours, 100%) cells. (F) Measurement of pSer2-RNAP2, BCL2A1 mRNA and Bfl-1 protein levels in OCILY10 cells at the indicated time points following 0.1 µM AZD4573 treatment. (G) Eµ-Myc lymphoma cells ectopically expressing BCL2A1, MCL1, BCL2, BCL2L1, or BCL2L2 prosurvival genes were treated with the CDK9 inhibitor AZ’5576 at the indicated concentrations for 24 hours. Apoptosis was measured by flow cytometric annexin V staining.
We next performed cycloheximide (CHX) chase experiments, with or without addition of the proteasome inhibitor MG132 to confirm rapid, proteasome-mediated depletion of both Bfl-1 and Mcl-1 (Figure 4D; supplemental Figure 4D-E). In all 3 models evaluated, Bfl-1 and Mcl-1 exhibited half-lives under 1 hour, whereas other prosurvival proteins exhibited greater stability with half-lives longer than 9 hours (Figure 4E; supplemental Figure 4F-G). To confirm CDK9 inhibitor mediated depletion of Bfl-1 occurred from direct transcriptional suppression of BCL2A1, a timecourse of the in vitro pharmacodynamic response to AZD4573 in the OCILY10 model was assessed. Consistent with previous results,16 pSer2-RNAP2 was reduced by 50% within 0.5 hours after AZD4573 treatment, which triggered a time-dependent reduction in BCL2A1 transcript levels 2 hours after dosing, followed by an equivalent loss of protein by 4 to 6 hours, which aligns with the aforementioned half-life of Bfl-1 (Figure 4F). Additionally, in transgenic Eµ-MYC lymphoma cells overexpressing BCL2A1 or MCL1, the selective, probe CDK9 inhibitor, AZ’5576, partially induced apoptosis relative to the more robust signal in the parental controls but completely failed to do so in the clones expressing the more stable prosurvival genes (Figure 4G). This provided additional support for the ability to target Bfl-1 via acute CDK9 inhibition.
Bfl-1 levels predict CDK9 and Mcl-1 inhibitor antitumor responses across DLBCL cell line and PDX tumors in vivo
The differential apoptotic response between CDK9 and Mcl-1 inhibitors in Bfl-1+ lymphomas implicated Bfl-1 levels as a predictive biomarker for antitumor responses to these respective agents in vivo. To test this hypothesis, mice harboring subcutaneous DLBCL xenografts were dosed with AZD4573 or AZD5991 using clinically relevant intermittent dosing schedules. Consistent with the in vitro phenotype, transient CDK9 inhibition upon AZD4573 administration resulted in robust tumor regressions in Bfl-1–expressing OCILY10 and TMD8 xenografts whereas treatment with AZD5991 achieved only a partial antitumor response (Figure 5A-B). AZD4573-mediated antitumor activity in OCILY10 tumor-bearing mice was associated with pharmacodynamic reductions of pSer2-RNAP2, Bfl-1, Mcl-1 and an increase in caspase-3 cleavage (Figure 5C) with no evidence of drug-induced body weight loss (supplemental Figure 5A-B).
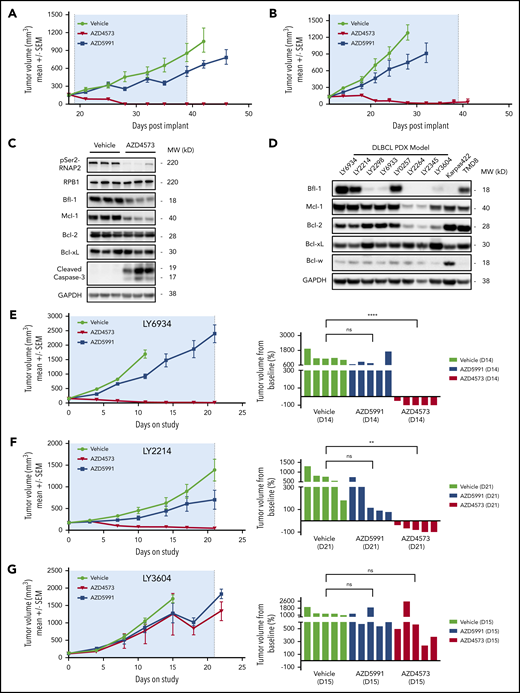
Bfl-1 is a positive response biomarker for CDK9 inhibitor antitumor efficacy across DLBCL cell line and PDX tumors in vivo. (A-B) CB17 SCID mice were implanted with OCILY10 (A) or TMD8 (B) cells and xenograft tumors grew to ∼200 mm3 prior to treatment with AZD4573 or AZD5991. (C) Pharmacodynamic analysis of pSer2-RNAP2, cleaved caspase-3, and antiapoptotic Bcl-2 family proteins in OCILY10 xenograft tumors after 4 hours of AZD4573 treatment. Western blot loading control, GAPDH. (D) Western blot analysis of prosurvival Bcl-2 family protein levels across 8 DLBCL PDX tumor models. Loading control, GAPDH. (E-G) NOD SCID mice were implanted with LY6934 (E), LY2214 (F), or LY3604 (G) DLBCL patient-derived tumor cells and treated with either AZD4573 or AZD5991 for 3 dosing cycles. Left panels, antitumor drug responses throughout the entire study duration. Right panels, individual animal tumor volumes at the indicated day. **P < .01; ****P < .0001. ns, no significance.
Bfl-1 is a positive response biomarker for CDK9 inhibitor antitumor efficacy across DLBCL cell line and PDX tumors in vivo. (A-B) CB17 SCID mice were implanted with OCILY10 (A) or TMD8 (B) cells and xenograft tumors grew to ∼200 mm3 prior to treatment with AZD4573 or AZD5991. (C) Pharmacodynamic analysis of pSer2-RNAP2, cleaved caspase-3, and antiapoptotic Bcl-2 family proteins in OCILY10 xenograft tumors after 4 hours of AZD4573 treatment. Western blot loading control, GAPDH. (D) Western blot analysis of prosurvival Bcl-2 family protein levels across 8 DLBCL PDX tumor models. Loading control, GAPDH. (E-G) NOD SCID mice were implanted with LY6934 (E), LY2214 (F), or LY3604 (G) DLBCL patient-derived tumor cells and treated with either AZD4573 or AZD5991 for 3 dosing cycles. Left panels, antitumor drug responses throughout the entire study duration. Right panels, individual animal tumor volumes at the indicated day. **P < .01; ****P < .0001. ns, no significance.
In addition to the subcutaneous cell-line xenograft models, we also characterized 8 DLBCL PDX tumors for Bcl-2 family antiapoptotic levels and observed a similar bimodal distribution of Bfl-1 protein levels that is comparable to the cell-line expression pattern (Figure 5D). Using 2 PDX models with Bfl-1 protein levels equivalent to or greater than the TMD8 cell line, intermittent dosing of AZD4573 in models LY6934 and LY2214 produced regressions whereas AZD5991 exhibited minimal efficacy throughout the 21-day dosing period (Figure 5E-F). Conversely, 2 models with low or no Bfl-1 protein expression, LY3604 and LY2345, exhibited minimal tumor growth inhibition in response to either CDK9 or Mcl-1 inhibition (Figure 5G; supplemental Figure 5C). Taken together, these results underscore that the level of Bfl-1 expression is critical for predicting in vivo lymphoma tumor responses to CDK9 or Mcl-1 inhibitor treatment.
Discussion
In multiple combination regimens, venetoclax is currently approved for the treatment of select leukemic malignancies and is registered in over 100 ongoing clinical trials, demonstrating extensive anticancer therapeutic potential for BH3 mimetics.29 Across hematological cancer cell-line panels Bcl-2 and Mcl-1 inhibitors display enhanced antitumor potency in leukemia and myeloma-derived neoplasms compared with lymphomas.9,11,23 Consistent with the preclinical data, venetoclax monotherapy treatment in 106 non-Hodgkin lymphoma patients produced an inferior overall response rate (44%)30 compared with that observed for 116 CLL patients (79%).2 To date, however, clinical response data for Mcl-1 inhibitors has not been reported. In the present study, we identified that Bfl-1 expression in preclinical lymphoma models reduces the apoptogenic potential of Bcl-2 and Mcl-1 inhibitors while Bfl-1 suppression sensitizes these lymphomas to BH3 mimetics in both monotherapy and combination settings. Although this is a promising therapeutic vulnerability for certain lymphomas, there are currently only stapled peptides and early-stage small molecules targeting Bfl-1, but none are in clinical development.31-34 However, in this body of work, we identified CDK9 inhibitors as a novel modality to target Bfl-1. Intermittent dosing of the selective inhibitor AZD4573 results in acute CDK9 inhibition and subsequent transient transcriptional suppression that exploits the labile nature of both Bfl-1 and Mcl-1 to activate intrinsic apoptosis. In codependent preclinical lymphoma cell lines and DLBCL PDX models, AZD4573 treatment generated a rapid apoptogenic response in vitro and resulted in tumor regressions in vivo, contrasting with the lack of response to AZD5991, venetoclax, or the combination of the two.
Prior studies using first-generation CDK9 inhibitors, like flavopiridol, have reported alternative CDK9-dependent antitumor mechanisms of action that are distinct from the canonical apoptogenic phenotype arising from depletion of Bfl-1 and/or Mcl-1.35,36 Notably, these mechanisms (eg, MYC modulation17 ) are elucidated using continuous inhibition of CDK9 for multiple days, which may downregulate additional genes critical for normal cellular processes. Similarly, initial clinical studies that evaluated frequent administration of flavopiridol reported narrow therapeutic margins from patients experiencing multiple adverse events. Consequently, these lengthy infusions are no longer being investigated.37,38 Rather, we demonstrated how transient and selective CDK9 inhibition upon acute exposure to AZD4573 was sufficient to suppress Bfl-1 and Mcl-1 to trigger a rapid and robust apoptogenic response. The intermittent clinical-dosing regimen of AZD4573, currently in clinical development (NCT03263637), was predicted to reflect the preclinical exposure that aims to maximize the therapeutic window by rapidly inducing cell death while allowing sufficient recovery from potential on-target toxicities in the intervening time.16 Thus, transient CDK9-mediated inhibition of Bfl-1 and Mcl-1 and resulting rapid induction of apoptosis may be more clinically translatable than alternative antitumor mechanisms requiring prolonged inhibition of CDK9.
Preliminary research suggests suppressing Bfl-1 in addition to Mcl-1 should not add significant safety liabilities. Since homozygous loss of BCL2A1 results in only small decreases in certain T-cell subpopulations and conventional dendritic cells, Bfl-1 is likely not essential for the development and survival of most normal, healthy tissue.39 This benign phenotype is likely attributed to the restricted, bimodal expression of Bfl-1 across primary tissue, as well as hematological cancers, which stands in stark contrast to Bcl-xL and Mcl-1 that exhibit higher and more ubiquitous expression. Expectedly, homozygous deletion of BCL2L1 or MCL1 in mice is embryonic lethal,40,41 which aligns with the clinical observations of on-target, dose-limiting toxicities upon pharmacological inhibition of Bcl-xL.42 Venetoclax, however, has a very manageable toxicity profile,2,3,30 and the expression pattern of Bcl-2 is similar to that of Bfl-1 in primary tissues (supplemental Figure 1D). Therefore, targeting Bfl-1 to combat emerging dependencies on this prosurvival protein may yield acceptable therapeutic margins.
Multiple reports have associated Bfl-1 overexpression with acquired venetoclax resistance in hematological cancers. Venetoclax drug-tolerant mantle cell lymphoma cells with 18q21 amplicon loss express functional BCL2A1.43 CLL cells also become resistant to Bcl-2 inhibition upon CD40 stimulation; however, in addition to Bfl-1, Mcl-1 and Bcl-xL are similarly upregulated, making it difficult to deconvolute functional dependencies.44,45 Our current work characterizing the endogenous role of Bfl-1 in lymphoma cell lines revealed a high proportion of models harboring constitutive activation of NF-κB and NPM-ALK, which each bind the BCL2A1 promoter to induce expression.46,47 These cell lines were characterized as codependent on Bfl-1 and another antiapoptotic protein for survival. Interestingly, Mcl-1 inhibitors were profoundly more effective at inducing robust apoptotic responses upon Bfl-1 suppression compared with Bcl-2 or Bcl-xL inhibitors. These results aligned with the significant increase in Bim in complex with Mcl-1 upon Bfl-1 suppression compared with the minor and less frequent increase in Bim in complex with Bcl-2. Importantly, we also observed enhanced complex formation between Mcl-1 and Noxa, which exclusively binds Bfl-1 and Mcl-1 and may further elicit the codependency of Bfl-1+ lymphomas on Mcl-1 to evade apoptosis.33,48
Recent reports also associate BCL2A1 levels with insensitivity to venetoclax in leukemias. A retrospective analysis of 100 AML patients treated with azacytidine plus venetoclax revealed patient tumor cells classified as monocytic-like (FAB-M5) were highly refractory to the combination therapy and overexpressed BCL2A1 and MCL1 antiapoptotic genes.49 Furthermore, BCL2A1 was the top apoptotic gene correlating with venetoclax resistance from over 200 treated primary AML samples ex vivo, many of which were also classified as FAB-M5.50 However, our internal leukemia cell lines did not intrinsically express Bfl-1 protein by western blot (supplemental Figure 1B), although ectopic expression of Bfl-1 in the FAB-M5 MV411 AML model effectively reduced the apoptogenic response to AZD5991, suggesting the presence of Bfl-1 in this context promotes cell survival. Interestingly, AZD4573 remained highly effective at activating apoptosis in this model (Figure 3E), suggesting the posttranslational elements promoting rapid Bfl-1 turnover in lymphomas are similarly present in other hematological malignancies. Importantly, our cell lines were cultured without bone marrow–enriched proinflammatory cytokines like IL-8, GM-CSF, and TNF-α, which have been reported to induce Bfl-1 through NF-κB activation.51-53 Therefore, leukemic tumors localized in the bone marrow or that intrinsically harbor an inflammatory transcriptome54 (eg, chronic myelomonocytic leukemia) may express Bfl-1 to enhance cell survival by redistributing mitochondrial dependencies on multiple antiapoptotic proteins.
The correlation between AZD4573-mediated antitumor responses and Bfl-1 expression in DLBCL PDX models supports using Bfl-1 as a patient selection biomarker for clinical-stage CDK9 inhibitors. However, practically measuring Bfl-1 levels in hematological cancer patients with circulating disease remains challenging, as we could not successfully validate commercial Bfl-1 antibodies compatible with immunophenotyping techniques like flow cytometry (data not shown). Therefore, more work should be invested into developing additional Bfl-1 antibodies, or exploring antibody-independent methods, including single cell RNA-sequencing55 as a surrogate for Bfl-1 protein abundance, considering the positive correlation for BCL2A1/Bfl-1 mRNA and protein levels (Figure 1C).
In summary, we have demonstrated that selective CDK9 inhibition downmodulates both Bfl-1 and Mcl-1 to drive striking antitumor activity in preclinical lymphoma models that require inhibition of both antiapoptotic proteins to induce apoptosis. Because BH3 mimetics targeting Bfl-1 have yet to enter clinical development, intermittent dosing of clinical-stage CDK9 inhibitors, such as AZD4573, could provide a therapeutic option for patients with DLBCL or other lymphomas that express Bfl-1. Likewise, CDK9 inhibitors could be useful tools in the arsenal to combat the Bfl-1-overexpressing segment of an emerging and complex venetoclax resistance landscape.44 This work supports further exploration of CDK9-inhibitor effectiveness, as monotherapy or in combination, in other malignancies expressing high levels of Bfl-1, including solid tumors.56,57
Raw data can be made available upon request by e-mailing justin.cidado@astrazeneca.com.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Haley Woods and Haiyun Wang for developing MSD immunoassays to quantify Mcl-1:Bim and Bcl-2:Bim complex levels. The visual abstract was created using BioRender.com.
The contributions from G.P.G. and R.W.J. were supported by grants from the Cancer Council Victoria, National Health and Medical Research Council Australia, The Peter MacCallum Cancer Foundation, and The Kids Cancer Project. AZ’5576 was provided to G.P.G. and R.W.J. under MTA agreement with AstraZeneca.
Authorship
Contribution: S.B., T.P., L.D., and J.C. contributed to study conception and design; S.B., L.D., and J.C. wrote the manuscript; S.B., T.P., M.S.M., G.P.G., M.M.W., N.A., M.H., and J.C. acquired and analyzed data; S.B., M.H., W.S., J.C.S., R.W.J., S.E.F., L.D., and J.C. provided study supervision; and all authors revised and edited the manuscript.
Conflict-of-interest disclosure: S.B., T.P., M.S.M., M.M.W., N.A., M.H., W.S., J.C.S., S.E.F., L.D., and J.C. are employees for AstraZeneca. R.W.J. has received research support from AstraZeneca, Roche, Merck, and Bristol Myers Squibb. G.P.G. is a member on Roche, Novartis, Gilead, and Janssen Pharmaceutical advisory boards. R.W.J. is a paid consultant and shareholder in MecRx.
Correspondence: Justin Cidado, AstraZeneca, 35 Gatehouse Dr, Waltham, MA 02451; e-mail: justin.cidado@astrazeneca.com.
