

In this issue of Blood, 1 describe a Toll-like receptor 9 (TLR9)-driven mechanism of therapeutic escape/evasion in patients with chronic lymphocytic leukemia (CLL), mediated by synergistic survival signaling via nuclear factor (NF)-κB and STAT3. These findings suggest a potential CLL treatment strategy using combined targeting of TLR9 and Bruton's tyrosine kinase (BTK).
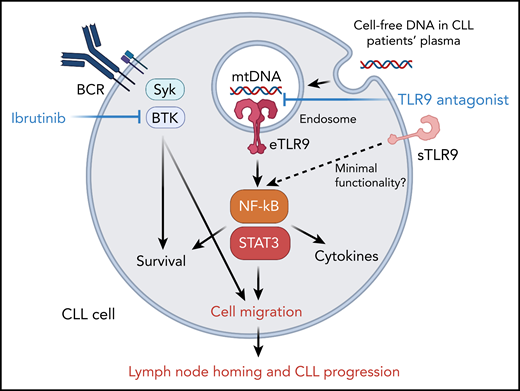
The endogenous TLR9 signaling supports CLL cell migration, homing to lymphoid organs and progression. Cell-free and mitochondrial DNA, with unmethylated cytosine-phosphate-guanosine motifs, triggers activity of TLR9 in CLL cells. TLR9 activation stimulates NF-κB and STAT3 signaling, which synergize with BTK to promote CLL cell migration and homing into lymphoid organs.
The endogenous TLR9 signaling supports CLL cell migration, homing to lymphoid organs and progression. Cell-free and mitochondrial DNA, with unmethylated cytosine-phosphate-guanosine motifs, triggers activity of TLR9 in CLL cells. TLR9 activation stimulates NF-κB and STAT3 signaling, which synergize with BTK to promote CLL cell migration and homing into lymphoid organs.
CLL is one of the most common adult leukemias, and the incidence and prevalence of CLL is increasing. Therapeutic options for CLL patients have expanded in recent years providing for more personalized regimens using targeted agents, such as inhibitors of BTK, phosphatidylinositol 3-kinase δ/γ, or B-cell lymphoma 2 signaling downstream from B-cell receptor (BCR). Despite these advances, must unmutated CLL (U-CLL) remains incurable except for allogenic stem cell transplantation.2 Beyond BCR signaling, TLR9 has been long recognized for contributing to CLL cell activity. TLR9 is an innate immune receptor and an endosomal sensor of pathogenic DNA or mitochondrial DNA released from dying cells. Synthetic TLR9 agonists, such as oligodeoxynucleotides containing unmethylated cytosine guanine motifs (CpG ODN), have been explored for CLL therapy based on early results demonstrating that CpG ODNs can induce CLL cell apoptosis and immune activation.3 However, the follow-up studies revealed a dichotomy in CLL responses to TLR9 agonists that also induced undesirable effects, such as stimulating CLL proliferation, suppressing of T-cell activity, and promoting therapeutic resistance.4 The reasons for the multiplicity of TLR9 roles is only partly understood, but they seem related to CLL genetics defining the interplay between TLR9 and BCR signaling. For example, ZAP-70 tyrosine kinase expressed mainly in U-CLL and not in mutated CLL (M-CLL) is necessary for bridging TLR9 to BCR signaling to promote U-CLL cell survival.5
In this study, Kennedy et al focused on the underappreciated role of endogenous TLR9 triggering progression of CLL and the potential therapeutic opportunities for interfering with this signaling (see figure). Analyzing plasma samples from 37 patients with CLL, the authors found a correlation between the elevated cell-free DNA (cfDNA) levels and markers of poor prognosis, such as white blood cell count, lymphocyte doubling time, and CD38 expression. Patients with CLL that have high median cfDNA levels showed significantly shorter time to first treatment. Compared with aged-matched healthy subjects, CLL patients also showed more than 28 times higher plasma levels of mitochondrial DNA, a well-described endogenous TLR9 trigger released from dying cells. This novel observation suggested that TLR9 drives autocrine signaling that supports CLL progression. In fact, CLL cells incubated in vitro with patient-derived plasma or with a synthetic TLR9 ligand (CpG ODN) showed TLR9-dependent induction of NF-κB/p65 and STAT3, 2 critical mediators of cancer cell survival and immune evasion. NF-κB/p65 and STAT3 signaling correlated with the upregulation of CD38, CD49d, and CD69 activation markers on CLL cells. Further immunophenotyping analysis assessed the intracellular expression of TLR9 in patients’ CLL cell populations. Although all CLL specimens were TLR9 positive, the activated CD38- and CD49d-positive CLL subsets showed the highest levels of TLR9 together with the activation of NF-κB/p65 and STAT3. Interestingly, the authors observed that activated CLL cells expressed TLR9 not only intracellularly, within endosomes but also on the cell surface. As expected, the surface TLR9 (sTLR9) had limited functionality, most likely because of the insufficient receptor processing. Previous studies demonstrated that to gain full activity TLR9 requires proteolytic cleavage, occurring specifically in the endolysosomes.6 Nonetheless, as suggested by the authors, the presence of sTLR9 provides a convenient marker of endogenous TLR9 signaling that has potential prognostic value. As evidenced by gene expression profiling, high levels of sTLR9 expression in CLL cells correlated with enrichment of transcriptional signatures of increased metabolic activity, lymphocyte activation/migration, and inflammation, with significant presence of NF-κB and STAT3 target genes.
Earlier studies implicated TLR9 signaling in promoting CLL cell survival through antigen stimulation and release of inflammatory or immunomodulatory cytokines.4,5,7 The authors here focused on the TLR9 effect on cell migration because leukemic cell trafficking into lymph node microenvironment correlates with the aggressiveness of CLL.4 In vitro experiments confirmed that TLR9 stimulation doubled the ability of primary CLL cells to migrate. In contrast, TLR9 antagonistic oligonucleotide decreased CLL cell migration and migration was further limited by a combination with BTK inhibitor (ibrutinib), consistently with the notion of BCR and TLR9 partnership in CLL cells. The reduced CLL migration correlated with the inhibition of NF-κB/p65 and STAT3, which are known regulators of cancer cell motility. When cultured in a hollow fiber bioreactor system, 9 of 10 tested primary CLL cells separated into circulating and migrating subsets with a clear upregulation of sTLR9 expression in the latter. Correspondingly, higher sTLR9 expression in lymph node–derived CLL cells compared with peripheral blood CLL cells was found in the matched patient samples. Finally, studies in xenotransplanted mouse models of primary CLL with high or low TLR9 levels demonstrated that leukemic cells expressing high TLR9 levels have dramatically improved splenic engraftment.
The report by Kennedy et al provides important new evidence of the role of endogenous TLR9 signaling in CLL biology with potential therapeutic implications. It is conceivable that such 2-pronged therapeutic intervention would effectively disrupt the partnership between BCR/BTK and TLR9 in driving CLL aggressiveness and therapeutic resistance. More in-depth studies will be needed to explore further the combination of TLR9 antagonist and BTK inhibitor in vivo or their efficacy against U-CLL and M-CLL subtypes, which differ in their dependance on BCR signaling.5 Nonetheless, the attractiveness of double-hit (BTK/TLR9) therapeutic strategy is also supported by a very recent report that targeting IRAK4, a kinase downstream from TLR9, synergizes with the cytotoxic effects of ibrutinib against CLL.8 The oligonucleotide approach used by the authors has the strong advantage of being selective to myeloid and B-cell lineages, which can spontaneously internalize ODNs, thereby limiting off-target effects.9 The TLR9 antagonist ODN used in the study has yet to be optimized for clinical application in terms of the pharmacokinetic/pharmacodynamic properties. However, given the predominantly negative and CLL promoting effects of endogenous TLR9 signaling, TLR9 blockade could reduce leukemia progression with minimal negative effects on the immune system activity against CLL. With TLR9 being commonly expressed in other hematologic malignancies, such as myeloid leukemia and B-cell lymphoma, findings from this study have broader therapeutic implications beyond CLL.
Conflict-of-interest disclosure: M.K. serves on the advisory board of Scopus Biopharma.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal