

In this issue of Blood, demonstrate that in MYC-driven B-cell lymphoma, activation of the ribosome biogenesis (RiBi) checkpoint triggers an apoptotic response, through the p53-induced, proteasome-dependent degradation of MCL-1.1
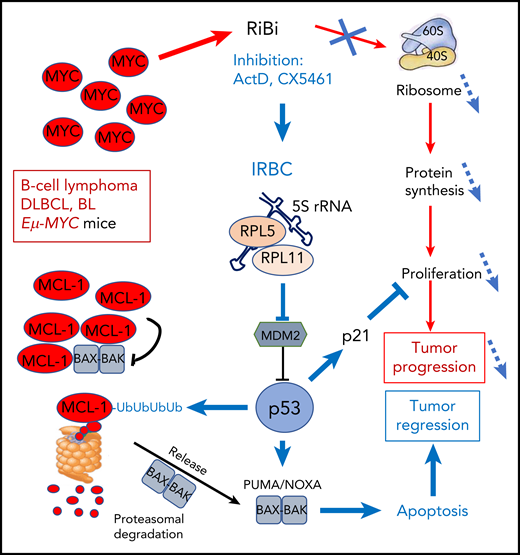
The amplified MYC oncogene in DLBCL and BL lymphoma strongly stimulates RiBi that supports cell proliferation and tumor progression through enhanced protein synthesis. Blocking RiBi with ActD or the pol-I inhibitor CX5461 decreases ribosome numbers impacting abnormal proliferation. In parallel, the IRBC comprising 5S rRNA, RPL5, and RPL11 is formed that blocks mdm2 to activate p53. p53 interferes with proliferation through induction of p21 and stimulates ubiquitination of the antiapoptotic BCL-2 family member MCL-1 that is frequently amplified in MYC-driven lymphomas. Ubiquitinated MCL-1 is then degraded in the proteasome to free BAK-BAX dimers that trigger mitochondrial membrane disruption and apoptosis, leading to tumor regression.
The amplified MYC oncogene in DLBCL and BL lymphoma strongly stimulates RiBi that supports cell proliferation and tumor progression through enhanced protein synthesis. Blocking RiBi with ActD or the pol-I inhibitor CX5461 decreases ribosome numbers impacting abnormal proliferation. In parallel, the IRBC comprising 5S rRNA, RPL5, and RPL11 is formed that blocks mdm2 to activate p53. p53 interferes with proliferation through induction of p21 and stimulates ubiquitination of the antiapoptotic BCL-2 family member MCL-1 that is frequently amplified in MYC-driven lymphomas. Ubiquitinated MCL-1 is then degraded in the proteasome to free BAK-BAX dimers that trigger mitochondrial membrane disruption and apoptosis, leading to tumor regression.
The MYC bHLH (basic Helix-Loop-Helix) transcription factor may be the most deregulated oncogene in all of human cancers, where it promotes increased cell metabolism and growth, enhanced survival, and abnormal proliferation.2 In particular, MYC is a strong stimulator of RiBi and is unique by its simultaneous enhancing effect on the activity of the RNA polymerases pol-I, pol-II, and pol-III. They respectively enhance transcription of the 47S precursor ribosomal RNA (rRNA), of messenger RNAs (mRNAs) for the 80 ribosomal proteins (RPs), and of the 5S rRNA, in order to boost the production of ribosomes.3
MYC is the hallmark oncogene amplified in aggressive diffuse large B-cell lymphoma (DLBCL) and Burkitt lymphoma (BL). Myc-driven lymphomas can be modeled in Eµ-MYC mice, where MYC is overexpressed in B lymphocytes under the control of the strong immunoglobulin heavy chain enhancer. Not surprisingly, it was observed that Eµ-MYC lymphomas are addicted to RiBi and protein synthesis. Indeed, the survival of Eµ-MYC mice was greatly increased by inducing a haploinsufficiency of either of the genes coding for the L24/RPL24 or L38/RPL38 RPs, correcting the enhanced protein synthesis rate of Eµ-MYC lymphoma cells back to homeostatic values.4 These results were in accordance with the ribosome being considered an active player in cancerous transformation and also an attractive anticancer target. Blocking either RiBi or the ribosome directly has shown promising effects5 and homoharringtonine, a protein synthesis inhibitor that directly blocks the ribosome, has been approved for clinical use in resistant chronic myeloid leukemia and is being evaluated in acute myeloid leukemia in various clinical trials. In a different direction, the depletion of the RPL24 gene triggers a p53-dependent cell-cycle arrest.6 Moreover, a multimolecular complex, the impaired ribosome biogenesis checkpoint (IRBC), consisting of 5S rRNA, RPL5, and RPL11, was then identified as a checkpoint of efficient RiBi. Upon RiBi inhibition, 5S rRNA, RPL5, and RPL11 are released from the preribosomal complex to form the IRBC that interacts with and blocks HDM2/mdm2 to induce p53 stabilization, acting as a tumor suppressor.7
The current study addresses the contribution of decreased global translation vs activation of IRBC/p53 in the anticancer effects of targeting RiBi. The authors used cell lines derived from Tp53+/+; Eµ-MYC mice. They show that the inducible short hairpin RNA-depletion of RPL7a or RPL11 both decreased protein synthesis. However, only the depletion of RPL7a, which does not affect the IRBC, could trigger a p53-dependent apoptotic response.
The MCL-1 antiapoptotic protein of the BCL-2 family is frequently coamplified with MYC to cooperate for transformation. Domostegui et al showed that after interference with RiBi, p53 induced polyubiquitination of MCL-1. Ubiquitinated MCL-1 is then degraded by the proteasome, leading to apoptotic cell death (see figure). This was not observed in a p53-mutated background.
The authors also show that low doses of actinomycin D (ActD), that blocks the transcription of rDNA by RNA polymerase I, induced IRBC and apoptosis of p53+/+ lymphoma cells. Their analysis of the Genomics of Drug Sensitivity in Cancer database revealed that cancer cell lines originating from blood cells are more sensitive to ActD than other tumor types, and among them the p53 status is predictive of the response to ActD. This was confirmed in vitro on a panel of BL and DLBCL cell lines. This important result supports the potential use of ActD in treating wt-TP53 hematological malignancies. ActD is already used, albeit at higher doses, for some pediatric tumors such as Wilms tumors, rhabdomyosarcoma, and Ewing sarcoma. Interestingly, it has been realized that interference with RiBi was a property of many classical antineoplastic drugs, such as anthracyclines.8 Also, oxaliplatin was recently shown to act through a RiBi stress response, in contrast to the other members of the cisplatin drug family that induce DNA damage.9 It would be interesting to explore the implications of this study on IRBC on the mode of action of these classical drugs.
Genetic defects that alter RiBi or ribosome function/production are at the roots of ribosomopathies, such as Diamond-Blackfan anemia or Schwachman-Diamond syndrome, that are congenital diseases associated with a high risk of developing cancer, particularly leukemia. One of the hypotheses to explain the malignant transformation is that the defects in RiBi/ribosome trigger during a hypoproliferative stage associated with inefficient protein synthesis, a salvage activation of p53 in an attempt to eliminate defective cells. However, the occurrence of a secondary genetic event that blocks the p53 pathway allows the cells to escape apoptotic elimination and to proceed to malignant transformation.10 Here also it could be interesting to analyze the potential role of IRBC and of MCL-1 in the support of the survival of RiBi defective cells. Finally, the work of Domostegui et al suggests that direct interference of MCL-1 function with specific BH3 mimetics could be an alternative approach in treating p53 mutated lymphomas, in association with RiBi inhibition.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal