

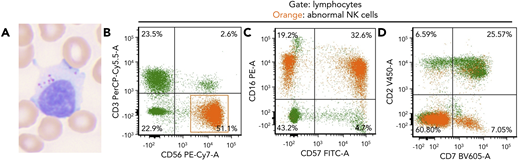
An 82-year-old woman presented with a history of lung and breast cancer, both treated surgically, and persistent asymptomatic lymphocytosis for 8 years. Peripheral blood showed increased large granular lymphocytes without other abnormalities (white blood cell count, 10.5 × 109/L; neutrophils, 47.8%; lymphocytes, 39.8%; monocytes, 8.6%; eosinophil, 3.2%; basophil, 0.6%) (panel A; Wright-Giemsa stain, original magnification ×60 objective, ×600 total magnification). Flow cytometry showed an abnormal natural killer (NK) cell population, 26% of leukocytes (2.7 K/μL), expressing CD56, CD16, subset CD57, minor subset CD7, and negative for CD3, CD2, and CD5 (panels B-D; orange). This NK population was identified 4 years prior and was thought to be due to a “reactive etiology.” Targeted mutational analysis (Stanford Actionable Mutation Panel for Hematopoietic and Lymphoid Malignancies) of the peripheral blood revealed TNFAIP3 p.R278fs, DNMT3A p.? (c.856-2A>G) and TP53 p.P177L mutations with variant allele frequencies of 8%, 22%, and 4%, respectively. Mutations in STAT3 or STAT5B were not identified.
Chronic lymphoproliferative disorder of NK cells (CLPD-NKs) shares morphologic features with reactive NK proliferations; thus, molecular techniques are often required to make a definitive diagnosis. This case of CLPD-NKs with TNFAIP3 and DNMT3A mutations is the second reported case of CLPD-NKs with TNFAIP3 mutation and highlights that CLPD-NKs, like T-cell large granular lymphocytic leukemia (T-LGLL), can show coexisting myeloid-associated mutations. It is unclear if the coexisting DNMT3A and TP53 mutations are within myeloid or NK cells to suggest concurrent CLPD-NKs and clonal hematopoiesis of indeterminate potential, or CLPD-NKs with myeloid-associated mutations, both phenomena reported to occur in T-LGLL.
An 82-year-old woman presented with a history of lung and breast cancer, both treated surgically, and persistent asymptomatic lymphocytosis for 8 years. Peripheral blood showed increased large granular lymphocytes without other abnormalities (white blood cell count, 10.5 × 109/L; neutrophils, 47.8%; lymphocytes, 39.8%; monocytes, 8.6%; eosinophil, 3.2%; basophil, 0.6%) (panel A; Wright-Giemsa stain, original magnification ×60 objective, ×600 total magnification). Flow cytometry showed an abnormal natural killer (NK) cell population, 26% of leukocytes (2.7 K/μL), expressing CD56, CD16, subset CD57, minor subset CD7, and negative for CD3, CD2, and CD5 (panels B-D; orange). This NK population was identified 4 years prior and was thought to be due to a “reactive etiology.” Targeted mutational analysis (Stanford Actionable Mutation Panel for Hematopoietic and Lymphoid Malignancies) of the peripheral blood revealed TNFAIP3 p.R278fs, DNMT3A p.? (c.856-2A>G) and TP53 p.P177L mutations with variant allele frequencies of 8%, 22%, and 4%, respectively. Mutations in STAT3 or STAT5B were not identified.
Chronic lymphoproliferative disorder of NK cells (CLPD-NKs) shares morphologic features with reactive NK proliferations; thus, molecular techniques are often required to make a definitive diagnosis. This case of CLPD-NKs with TNFAIP3 and DNMT3A mutations is the second reported case of CLPD-NKs with TNFAIP3 mutation and highlights that CLPD-NKs, like T-cell large granular lymphocytic leukemia (T-LGLL), can show coexisting myeloid-associated mutations. It is unclear if the coexisting DNMT3A and TP53 mutations are within myeloid or NK cells to suggest concurrent CLPD-NKs and clonal hematopoiesis of indeterminate potential, or CLPD-NKs with myeloid-associated mutations, both phenomena reported to occur in T-LGLL.
For additional images, visit the ASH Image Bank, a reference and teaching tool that is continually updated with new atlas and case study images. For more information, visit http://imagebank.hematology.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal