


Abstract
The standard treatment of thrombotic antiphospholipid syndrome (APS) is lifelong oral anticoagulation with a vitamin K antagonist (VKA), generally warfarin. A minority of patients with APS rethrombose despite seemingly adequate anticoagulation. These patients are deemed anticoagulant refractory. The management of anticoagulant-refractory APS is largely empirical and extrapolated from other clinically similar situations. Further options include increased VKA anticoagulation intensity or alternative antithrombotic strategies, including low-molecular-weight heparin, fondaparinux, the addition of antiplatelet therapy, and consideration of vascular options. Patients with anticoagulant-refractory thrombotic APS may have APS-associated thrombocytopenia, which necessitates balancing the risk of recurrent thrombosis vs bleeding to achieve adequate anticoagulation. The multiple mechanisms involved in the generation of the thrombotic phenotype in APS suggest that anticoagulation alone may not control thrombosis. Thus, other modalities, including adjunctive treatment (hydroxychloroquine, statins, and vitamin D) for APS-related thrombosis, merit consideration, as do immunomodulatory therapy and complement inhibition. Patients with APS may have coexistent systemic lupus erythematosus, which adds to the complexity of managing their thromboembolic disease. However, with attention to detail and judicious application of the limited data, it is possible to minimize the morbidity resulting from anticoagulant-refractory thrombotic APS. Multicenter studies are required to guide the sequence of interventions and their comparative efficacy in patients with anticoagulant-refractory thrombotic APS.
Introduction
Antiphospholipid syndrome (APS) is characterized by thrombosis (arterial, venous, and microvascular) and/or pregnancy morbidity in association with persistent antiphospholipid antibodies (aPLs): ≥1 of lupus anticoagulant (LA), anticardiolipin antibodies (aCL), or anti–β2-glycoprotein I antibodies (aß2GPI), present on 2 occasions at least 12 weeks apart.1 Triple positivity denotes the presence of all 3 antibodies. Approximately 50% of thrombotic events are lower limb deep venous thromboses (DVTs) and pulmonary emboli, with strokes and transient ischemic attacks accounting for ∼30%.2 The overall prevalence of APS is estimated to be 50 per 100 000 of the population,3 with a female:male ratio of 5:1.2 Catastrophic APS (CAPS), which accounts for 1% of cases, is associated with mortality rates of 29%, 41%, and 75% with triple therapy (anticoagulation, corticosteroid and plasma exchange/intravenous immunoglobulin [IVIG]), other combinations, and none of those treatments, respectively.4 APS classification criteria are being updated.5
What is anticoagulant-refractory thrombotic APS?
Anticoagulant-refractory thrombotic APS can be broadly defined as breakthrough thrombosis on standard treatment (ie, anticoagulation with warfarin or an alternative vitamin K antagonist [VKA]) while at a therapeutic international normalized ratio (INR). In patients who rethrombose while at a therapeutic INR, it is important to ensure that the INR is not spuriously in target range because of interference caused by LA on thromboplastin.6,7 The majority of thromboplastins can be safely used in LA-positive patients; a thromboplastin insensitive to LA should be used for INR monitoring.6,7 Point-of-care INRs are variably affected by LA, and results must be interpreted with caution.7,8 Chromogenic factor X levels provide an LA-independent assessment of VKA intensity, although therapeutic ranges are not established.6,7,9 In situations in which the optimal target INR,10-13 or optimal antithrombotic strategy,12-14 is uncertain, as in APS-related stroke, it is challenging to determine precisely why a patient might rethrombose. Patients who rethrombose while on standard therapeutic-dose low-molecular-weight heparin (LMWH) should also be regarded as anticoagulant refractory. Suspected new thrombosis/thrombosis extension requires objective confirmation by using appropriate imaging.
Prevalence figures for anticoagulant-refractory thrombotic APS are lacking, although clinical experience suggests it is rare. There is some overlap with the catastrophic thrombotic syndromes (reviewed elsewhere15 ). Because of the paucity of evidence, the management of anticoagulant-refractory thrombotic APS is largely empirical. Treatment merits consideration of additional modalities, extrapolated from similar clinical situations in other prothrombotic disorders. The opinions expressed in this article reflect the authors’ practical experience in managing these challenging patients.
The annualized risk of recurrent thrombosis in patients with APS on standard-intensity VKA was 1.3% and 1.5% in 2 randomized controlled trials (RCTs), respectively16,17 : 4.3% in the Euro-phospholipid prospective cohort18 ; and 4.8% in a retrospective study on triple-positive patients.19 Direct oral anticoagulants (DOACs) are a potential alternative to VKAs; however, in the recent trials,20-22 some raised concern about DOAC-related rethrombosis.21,22 Results of TRAPS (Trial on Rivaroxaban in Thrombotic Antiphospholipid Syndrome) prompted a risk assessment that led to the European Medicines Agency recommendation against the use of DOACs for APS, especially in triple-positive patients.23 The place of DOACs in APS has not been established due to the lack of definitive evidence.24 The efficacy of DOACs has been confirmed vs standard-intensity warfarin in phase 3 trials in general population venous thromboembolism (VTE) and atrial fibrillation patients.25,26 However, the doses of DOACs used in these studies may not be effective in patients who rethrombose while receiving standard-intensity VKA.27 Based on the limited data available, DOACs are not recommended in patients with APS and recurrent thrombosis while within therapeutic range on standard-intensity VKA.13,24 It follows that DOACs should not be used in patients with anticoagulant-refractory thrombotic APS.
Synopsis of pathophysiology mechanisms in thrombotic APS
The precise mechanisms involved in aPL-related prothrombotic and proinflammatory changes are complex and have been discussed elsewhere.28,29 In brief, pathogenic aPL are believed to bind β2GPI, leading to exposure of a cryptic domain 1 Arg39-Arg43 epitope, in its more open oxidized form. This form lacks free thiols, which are increased during oxidative stress and may be raised in patients with APS.28-31 The aPL–β2GPI complex can then cause cross-linking to many surface receptors, with subsequent activation of effector cells, leading to release of prothrombotic and proinflammatory mediators. Figure 1 provides more detail about the individual receptors and molecules which are (mostly) upregulated as a consequence. Diverse cells, including endothelial cells, monocytes, neutrophils, and platelets, are involved in this process. Intracellular signaling, in particular through MAPKs and the key transcription regulator NF-κB, is integral in activating these target cells. Increasing evidence supports the concept that the complement system is involved in the pathogenesis of APS, with lower levels of both C3 and C4 and concomitant increases in C3a-desArg and C4a reported in patients with primary APS,32 suggesting complement activation. Aberrant activation of the coagulation cascade of serine proteases may be contributory, perhaps through antibodies that cross-react with serine proteases and factor Xa.29 Other possible contributory factors include enhanced tissue factor expression and impaired activation of protein C.33 The multiple mechanisms involved in the generation of the thrombotic phenotype in APS suggest that anticoagulation alone may not control thrombosis.
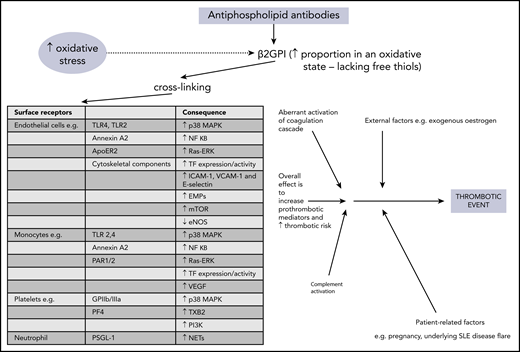
Overview of pathophysiology mechanisms in thrombotic APS. ApoER2, apolipoprotein E receptor 2; EMPs, endothelial microparticles; eNOS, endothelial nitric oxide synthase; GPIIb/IIIa, glycoprotein IIb/IIIa; mTOR, mammalian target of rapamycin; NETs, neutrophil extracellular traps; PAR, protease-activated receptor; PF4, platelet factor 4; PI3K, phosphatidylinositol 3-kinase; PSGL-1, P-selectin glycoprotein ligand 1; Ras-ERK, Ras-extracellular signal-related kinase; TF, tissue factor; TLR, toll-like receptor; TXB2, thromboxane B2; VCAM-1, vascular cell adhesion molecule 1; VEGF, vascular endothelial growth factor.
Overview of pathophysiology mechanisms in thrombotic APS. ApoER2, apolipoprotein E receptor 2; EMPs, endothelial microparticles; eNOS, endothelial nitric oxide synthase; GPIIb/IIIa, glycoprotein IIb/IIIa; mTOR, mammalian target of rapamycin; NETs, neutrophil extracellular traps; PAR, protease-activated receptor; PF4, platelet factor 4; PI3K, phosphatidylinositol 3-kinase; PSGL-1, P-selectin glycoprotein ligand 1; Ras-ERK, Ras-extracellular signal-related kinase; TF, tissue factor; TLR, toll-like receptor; TXB2, thromboxane B2; VCAM-1, vascular cell adhesion molecule 1; VEGF, vascular endothelial growth factor.
Case 1: anticoagulant-refractory recurrent VTE
A 28-year-old woman had a right popliteal vein DVT at 20 weeks gestation, treated with a standard therapeutic dose of LMWH during the remainder of the pregnancy. This was followed by a 3-month course of standard-intensity warfarin postpartum. The following year, she had a miscarriage at 11 weeks gestation. In her third pregnancy 1 year later, she received prophylactic-dose LMWH throughout pregnancy and for 6 weeks postpartum. The patient developed preeclampsia at 28 weeks gestation necessitating emergency Caesarean section, with delivery of a female infant (birth weight, 737 g).
At age 40 years, the patient had an unprovoked left popliteal vein DVT and received initial therapeutic-dose LMWH followed by standard-intensity warfarin. She had persistent triple-positive aPL, with high-titer aß2GPI at 40.7 (normal range [NR], 0-10) U/mL and medium immunoglobulin G (IgG) aCL at 26.6 (NR, 0-12), confirmed on retesting after 12 weeks. The plan was for life-long warfarin, and she was adherent to treatment. Four months later, the patient developed symptomatic DVT extension to the left common femoral vein, while the (venous) INR was therapeutic. She had bridging standard-treatment dose LMWH, and the target INR was increased to 3.5. Eight months later, further symptoms prompted a repeat duplex scan. This showed proximal thrombus extension within the common femoral vein also involving the external iliac vein. These developments occurred despite therapeutic high-intensity warfarin with good anticoagulant control, when the INR was 3.4. The warfarin was switched to high-intensity LMWH, ∼20% above standard dose, with split-dose dalteparin 10 000 units every 12 hours (weight, 83 kg). She developed a left sigmoid sinus thrombosis the following year, diagnosed on a computed tomographic venogram. She had cerebral angiography for a suspected dural arteriovenous fistula, not confirmed. Postangiogram, the patient had a small acute frontal embolic infarct. LMWH was escalated to ∼30% above standard dose; that is, dalteparin 12 500 and 10 000 units 12 hourly, with target peak anti–factor Xa levels 1.0 to 1.20 U/mL.
One year later, the patient had an unprovoked right middle lobe segmental pulmonary embolism. The LMWH was switched to fondaparinux 7.5 mg once daily (weight, 79 kg). She was also prescribed rituximab (375 mg/m2 × 4 weekly doses) and has not rethrombosed subsequently (for 5 years). She remains triple positive with the most recent IgG aß2GPI 6,100 (NR 0-10) U/mL and aCL 2,024 (NR 0-12) U/mL (by chemiluminescence). Vitamin D levels were noted to be insufficient when LMWH was started (36 [reference range, 25-125; insufficient, 25-50] nmol/L), with replacement treatment instituted. Bone mineral density and lipid status have remained normal throughout.
Comments about case 1
This patient has thrombotic and obstetric APS. Her history raises the critical question of whether patients with APS and recurrent/anticoagulant-refractory thrombosis can be identified early in their clinical course. Risk factors for this likely include a triple-positive phenotype19 (present in this patient), anti-β2GPI antibodies that bind to a limited epitope (Arg39-Arg43) on domain 1,34 and anti–protein C antibodies linked to acquired activated protein C resistance.35
The evolution of APS-related thrombosis is multifactorial. A variety of environmental or patient-related factors are implicated in VTE and may also contribute to APS-related recurrent thrombosis; these include age ≥40 years, obesity, immobility, trauma, surgery, exogenous estrogens, pregnancy,36 and systemic lupus erythematosus (SLE) and other autoimmune disease, discussed elsewhere.33 However, the role of these factors, or that of ethnicity, in APS-related VTE is not fully defined. Arterial thromboembolism is also increased in SLE.33 Active management of conventional cardiovascular risk factors in patients with APS is important, emphasized in a prospective study on the APS Antiphospholipid Syndrome Alliance for Clinical Trials and International Networking (ACTION) cohort. This report indicated that patients with APS (n = 379) with recurrent arterial, but not venous, thrombosis had higher adjusted global antiphospholipid syndrome scores (aGAPSS), which incorporate hyperlipidemia and arterial hypertension in addition to aPL (mean ± SD, 8.1 ± 2.9 vs 6 ± 3.9; P < .05).37 In patients with recurrent thrombosis, potential provoking factors for VTE, detailed earlier, and additional prothrombotic conditions, such as cancer38 or myeloproliferative neoplasm,39 should be considered (although not implicated in this case).
This case illustrates the progression of anticoagulation and additional therapeutic options for APS-related recurrent/anticoagulant-refractory thrombosis (Figure 2). Points to consider when rethrombosis occurs are summarized in Table 1. The recommended approach, following a first unprovoked VTE in general population patients who have a low or moderate bleeding risk, is extended anticoagulant therapy.40 A systematic review reported that a positive aPL test result seems to predict an increased risk of recurrence in patients with a first VTE; however, the available evidence was of very low quality.41 A subsequent prospective study suggested that aPL and raised D-dimer levels seem to be independent predictors of recurrence after a first unprovoked VTE.42 Lifelong anticoagulation with a VKA is standard treatment of thrombotic APS.10,12,13,24,43,44 The optimal anticoagulation duration after provoked aPL-associated VTE is undefined.
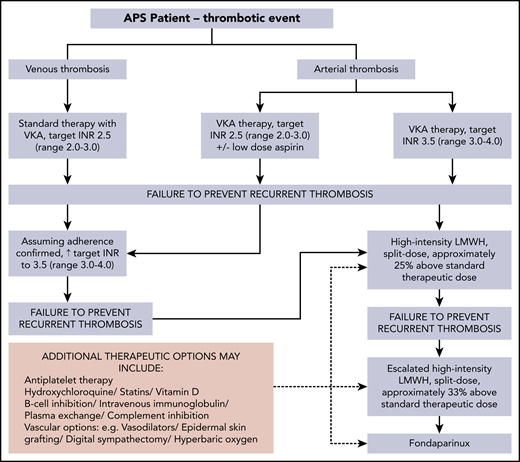
Progression of anticoagulation and additional therapeutic options for APS-related recurrent/anticoagulant-refractory thrombosis (authors’ approach).
Progression of anticoagulation and additional therapeutic options for APS-related recurrent/anticoagulant-refractory thrombosis (authors’ approach).
Factors to consider when assessing patients with suspected thromboembolism recurrence on standard-intensity warfarin/VKA
| 1. Confirmation by appropriate imaging of new thrombosis or thrombosis extension |
| 2. Review of the INR results before the thrombosis for assessment of patient adherence |
| 3. Check that the patient’s INR assessment has been performed by using an LA-insensitive thromboplastin |
| 4. Test for heparin-induced thrombocytopenia45 if rethrombosis occurs within 14 d of starting LMWH |
| 5. Consideration of provoking factors (eg, immobility, surgery) for VTE or additional risk factors for thrombosis (eg, malignancy, SLE or other autoimmune disease, myeloproliferative neoplasm) |
| 6. Consideration of bleeding risk factors (eg, gastrointestinal or uterine, thrombocytopenia), as such factors may limit anticoagulation intensity |
| 1. Confirmation by appropriate imaging of new thrombosis or thrombosis extension |
| 2. Review of the INR results before the thrombosis for assessment of patient adherence |
| 3. Check that the patient’s INR assessment has been performed by using an LA-insensitive thromboplastin |
| 4. Test for heparin-induced thrombocytopenia45 if rethrombosis occurs within 14 d of starting LMWH |
| 5. Consideration of provoking factors (eg, immobility, surgery) for VTE or additional risk factors for thrombosis (eg, malignancy, SLE or other autoimmune disease, myeloproliferative neoplasm) |
| 6. Consideration of bleeding risk factors (eg, gastrointestinal or uterine, thrombocytopenia), as such factors may limit anticoagulation intensity |
An appropriate anticoagulation plan should be guided by the INR result at the time of thrombosis. If the INR is subtherapeutic (<1.5 for target INR 2.0-3.0), warfarin/VKA may be resumed at standard intensity, with close INR monitoring. If rethrombosis occurs while at standard-intensity VKA, high-intensity VKA anticoagulation is widely used, although formal studies regarding this approach, as well as for an interim/bridging LMWH regimen, are lacking. Our practice is to stop the warfarin and start standard-intensity once-daily LMWH, continuing for 2 to 4 weeks, before reintroduction of warfarin with INR monitoring 2 to 3 times weekly. The LMWH dose is reduced by 50% when the INR rises to >2.0 and stopped when the INR is >2.7 and <3.0. An alternative to high-intensity VKA is the addition of aspirin; however, there is little evidence to indicate which approach is more effective.46
Rethrombosis while within a target INR range of 3.0 to 4.0, in our practice, leads to a switch to high-intensity LMWH, ∼20% above standard dose. Intravenous vitamin K, usually at a small dose (1-2 mg), is given to minimize bleeding risk. A switch back to the target INR range at which rethrombosis occurred may be considered after a reversible additional prothrombotic situation has resolved (eg, immobility).
High-intensity LMWH was used in this patient, as a recurrent DVT occurred despite therapeutic high-intensity VKA. This approach, in a retrospective study of 70 patients with cancer, resulted in no VTE recurrence in 91% and major bleeding in one. These patients, who had recurrent VTE while on oral anticoagulation, were either switched from VKA to LMWH (23 patients) or had their LMWH increased by 20% to 25% (47 patients). The follow-up was 3 months.47 We used escalated-dose high-intensity LMWH (∼30% above standard dose) after the unprovoked pulmonary embolus. This approach accords with the American College of Chest Physicians’ guideline recommendation of an increase of ∼25% to 33% for rethrombosis while on LMWH (grade 2C).40 In 2 small retrospective studies in patients with APS, in which 14 of 24 and 9 of 23 failed warfarin therapy, 1 and 3 patients, respectively, had recurrent thrombosis.48,49 Thus, LMWH offers an option in some patients who have rethrombosis on VKA.
Prolonged LMWH (between 3 and 24 months) is associated with a decrease in bone mineral density.50 Furthermore, 1α,25-dihydroxyvitamin D3 downregulates tissue factor and upregulates thrombomodulin expression in vitro.51 Vitamin D inhibits the expression of tissue factor in monocytes from patients with APS stimulated by aβ2GPI. Low vitamin D levels correlate with arterial/venous thrombosis in patients with APS.52 The 14th, 15th, and 16th International Congress on aPL Task Forces on APS Treatment Trends have recommended that vitamin D deficiency should be corrected, based on general population guidelines.13,43,44 We ensure that vitamin D levels are well into the normal range. These task forces also recommended consideration of hydroxychloroquine and statins in anticoagulant-refractory APS.13,43,44 Hydroxychloroquine, in studies in animal models and human aortic endothelial cells, improves procoagulant status and vascular function in APS by modulating endothelial nitric oxide synthase, leading to an improvement in the production of nitric oxide.53 It reduces the risk of thrombosis in patients with SLE and APS animal models54 and reportedly reduces aPL levels and arterial thrombosis recurrence in patients with primary APS.55 Statins have immunomodulatory, anti-inflammatory, and antithrombotic properties, in addition to their lipid-lowering effects.56 Fluvastatin reduces aPL-mediated tissue factor and monocyte adhesion to endothelial cells in vitro.57 A prospective open-label pilot study of fluvastatin for 3 months in 24 patients with APS reported reductions in proinflammatory and prothrombotic biomarkers, including interleukin-1β, vascular endothelial growth factor, tumor necrosis factor α, and soluble tissue factor.58
Fondaparinux was initiated in this patient after rethrombosis on escalated high-intensity LMWH. This synthetic analogue of heparin pentasaccharide, used mainly for the treatment of heparin-induced thrombocytopenia,45 has anti–factor Xa activity sevenfold higher than that of LMWH.59 There are few data about fondaparinux use in APS. One prospective study completed in 26 of 30 patients with DVT or pulmonary embolism, 13 each with hypercoagulable states (the number with APS was not clarified) and recurrent VTE, despite a therapeutic INR, showed no rethrombosis or major bleeding after 90 days of follow-up.60 Fondaparinux has also been used successfully in 2 patients with APS and microvascular thrombosis, in combination with mycophenolate mofetil (MMF).61 A recent report highlighted the use of fondaparinux in 3 patients with anticoagulant-refractory thrombotic APS, who remained thrombosis free over 40 months of follow-up.62 In vitro studies show no significant inhibition of osteoblast proliferation or activity with fondaparinux.63 Whether prolonged fondaparinux use is associated with preservation of bone mass is unknown. Consideration of inferior vena cava filters should be reserved for patients at high risk of pulmonary embolism when anticoagulation is contraindicated.64 However, an RCT of inferior vena cava filter placement in this situation showed no overall benefit.65
Not much is known about the use of rituximab in anticoagulant-refractory thrombotic APS. Four of 5 patients with SLE-related APS who had recurrent thrombosis on therapeutic warfarin had no rethrombosis after rituximab.66 The authors of this report and Ioannou et al67 described a notable drop in aPL positivity with rituximab use. However, this observation was not confirmed in the RITAPS (Rituximab in APS) phase 2 open-label prospective pilot study of rituximab for APS patients with noncriteria manifestions.68 Table 2 details non-anticoagulation options for potential use in anticoagulant-refractory thrombotic APS.
Non-anticoagulation options (generally empirical) for potential use in anticoagulant-refractory thrombotic APS
| When to consider | Options | Caveats and comments |
|---|---|---|
| Rethrombosis despite standard-intensity VKA: increase to high-intensity VKA | Vitamin D: correct vitamin D deficiency based on general population guidelines in patients with thrombotic APS | Theoretical reasons for using vitamin D are compelling, but clinical data are limited |
| Statins: correct hyperlipidemia/dyslipidemia based on general population guidelines | Good theoretical reasons for using statins but be mindful of myalgia (∼10%) and (rarely) myositis | |
| Hydroxychloroquine | Good theoretical support from animal models and some clinical data | |
| Very few side effects but long-term use warrants regular ophthalmic examination for rare “bullseye maculopathy” | ||
| Antiplatelet agents | Paucity of evidence for addition of low-dose aspirin to standard-intensity VKA vs switch to high-intensity VKA | |
| Associated with increased bleeding risk | ||
| Rethrombosis despite high-intensity VKA and subsequent high-intensity LMWH (∼20% above standard-therapeutic dose): increase to escalated-dose LMWH (∼30% above standard-therapeutic dose), then fondaparinux | B-cell inhibition | May be particularly useful if concomitant thrombocytopenia is present |
| Approximately 10% of patients with SLE develop hypogammaglobulinemia with consequent infection risk | ||
| IVIG | May be particularly useful if concomitant thrombocytopenia refractory to rituximab but also potentially prothrombotic; therefore, cautious dosing is advised | |
| Plasma exchange | Suggest replacement with 100% FFP to minimize coagulopathy and bleeding risk with anticoagulation | |
| SD-FFP preferable because of reduced potential for adverse events | ||
| Complement inhibition | May be beneficial in APS-related refractory microvascular thrombotic states, including thrombotic microangiopathy or chronic persistent microvascular thrombosis | |
| Vaccinate against meningococcal infections | ||
| Vasodilators | The potential benefit of sildenafil and iloprost may exceed their vasodilatory effect, possibly mediated by platelet function inhibition and endothelial stabilization | |
| Surgical interventions to achieve vasodilation: lumbar sympathectomy, digital sympathectomy, sacral nerve stimulation | The principle is to block/reduce sympathetic mediated vasoconstriction of arterioles permanently | |
| Epidermal grafting | Autologous skin grafting in which the epidermal layer of the skin is harvested from the donor site, then excised and transferred onto the wound | |
| Hyperbaric oxygen therapy | This may be an option for patients with dermal ulceration or tissue loss secondary to ischemic APS | |
| Inferior vena cava filters | Mounting evidence shows little benefit |
| When to consider | Options | Caveats and comments |
|---|---|---|
| Rethrombosis despite standard-intensity VKA: increase to high-intensity VKA | Vitamin D: correct vitamin D deficiency based on general population guidelines in patients with thrombotic APS | Theoretical reasons for using vitamin D are compelling, but clinical data are limited |
| Statins: correct hyperlipidemia/dyslipidemia based on general population guidelines | Good theoretical reasons for using statins but be mindful of myalgia (∼10%) and (rarely) myositis | |
| Hydroxychloroquine | Good theoretical support from animal models and some clinical data | |
| Very few side effects but long-term use warrants regular ophthalmic examination for rare “bullseye maculopathy” | ||
| Antiplatelet agents | Paucity of evidence for addition of low-dose aspirin to standard-intensity VKA vs switch to high-intensity VKA | |
| Associated with increased bleeding risk | ||
| Rethrombosis despite high-intensity VKA and subsequent high-intensity LMWH (∼20% above standard-therapeutic dose): increase to escalated-dose LMWH (∼30% above standard-therapeutic dose), then fondaparinux | B-cell inhibition | May be particularly useful if concomitant thrombocytopenia is present |
| Approximately 10% of patients with SLE develop hypogammaglobulinemia with consequent infection risk | ||
| IVIG | May be particularly useful if concomitant thrombocytopenia refractory to rituximab but also potentially prothrombotic; therefore, cautious dosing is advised | |
| Plasma exchange | Suggest replacement with 100% FFP to minimize coagulopathy and bleeding risk with anticoagulation | |
| SD-FFP preferable because of reduced potential for adverse events | ||
| Complement inhibition | May be beneficial in APS-related refractory microvascular thrombotic states, including thrombotic microangiopathy or chronic persistent microvascular thrombosis | |
| Vaccinate against meningococcal infections | ||
| Vasodilators | The potential benefit of sildenafil and iloprost may exceed their vasodilatory effect, possibly mediated by platelet function inhibition and endothelial stabilization | |
| Surgical interventions to achieve vasodilation: lumbar sympathectomy, digital sympathectomy, sacral nerve stimulation | The principle is to block/reduce sympathetic mediated vasoconstriction of arterioles permanently | |
| Epidermal grafting | Autologous skin grafting in which the epidermal layer of the skin is harvested from the donor site, then excised and transferred onto the wound | |
| Hyperbaric oxygen therapy | This may be an option for patients with dermal ulceration or tissue loss secondary to ischemic APS | |
| Inferior vena cava filters | Mounting evidence shows little benefit |
FFP, fresh frozen plasma; SD-FFP, solvent-detergent fresh frozen plasma.
Case 2: anticoagulant-refractory persistent microvascular thrombosis, recurrent VTE, arterial thrombosis, thrombocytopenia, and bleeding
This patient was diagnosed aged 18 years with SLE, characterized clinically by arthritis and membranous glomerulonephritis; she was treated successfully with intramuscular steroids and hydroxychloroquine. The patient had an unprovoked proximal lower limb DVT at 30 years of age; after initial LMWH, she was maintained on standard-intensity warfarin. She had triple aPL-positivity with high-titer IgG aβ2GPI and aCL, both >100, NR 0 to 10 and 0 to 12 U/mL, respectively. At 39 years of age, the patient had an early miscarriage. Over several months, she developed severe pain from ulcers in the right shin (biopsy results showed microvascular thrombosis) and dorsum of the right foot, and a blue right second toe. A computed tomographic angiogram revealed right dorsalis pedis artery occlusion and a small right common iliac artery mural thrombus. The platelet count, previously stable at >150 000/μL, was 44 000/μL, with a hemoglobin level of 77 g/L and no red blood cell fragmentation. Renal and hepatic function test results, lactate dehydrogenase levels, and ADAMTS13 activity were normal. Her SLE was serologically active, with raised anti–double-stranded DNA antibodies and reduced C3 at 0.80 (NR, 0.90-1.8) g/L, but clinically quiescent. The warfarin was switched to split treatment dose LMWH, together with intravenous methylprednisolone (IVMP), plasma exchange (PEX), and rituximab (375 mg/m2 × 4 weekly doses), with clinical improvement and platelet levels >100 000/μL.
Over the next 6 months, the patient was treated with LMWH, IVMP, PEX, rituximab, and iloprost. She had 75% split treatment dose LMWH during iloprost infusion, with the iloprost withheld when platelet counts were <75 000/μL, in view of its platelet-inhibitory effect. The skin ulcers became necrotic but were treated with antibiotics, debridement, and epidermal grafting, with good results. Fifteen months after her initial presentation with skin ulcers, the patient was admitted with Citrobacter bacteremia, with a platelet count of 9000/μL. Treatment included IVMP, PEX, IVIG, and platelet transfusions. She subsequently underwent terminalization of an ischemic right hallux, and fourth and fifth toes, followed by angioplasty, PEX, and iloprost. She developed a postoperative Vascath-associated acute left common iliac and external iliac vein DVT and received IVIG and platelet transfusions to maintain platelet counts of 40 to 50 000/μL to enable therapeutic anticoagulation. The addition of MMF did not improve the thrombocytopenia and was switched to eltrombopag. She received a 6-month course of eculizumab and hyperbaric oxygen therapy (HBOT; 50 sessions over 10 weeks), with healing/improvement in the surgical wounds and ulcers (Figure 3). The thrombocytopenia improved, with platelets generally maintained >50 000/μL.
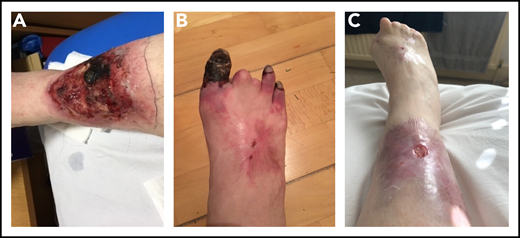
Photographs of right lower limb of patient 2 during disease progression. (A) Leg ulcer due to microvascular thrombosis (confirmed histologically). (B) Progressive gangrene in 3 toes, the 2 other toes having autoamputated. (C) Considerable improvement in the leg ulcer, following multimodal therapies, and the end result of the digital gangrene.
Photographs of right lower limb of patient 2 during disease progression. (A) Leg ulcer due to microvascular thrombosis (confirmed histologically). (B) Progressive gangrene in 3 toes, the 2 other toes having autoamputated. (C) Considerable improvement in the leg ulcer, following multimodal therapies, and the end result of the digital gangrene.
During the initial iloprost infusion, the patient’s platelet count dropped to 64 000/μL, and she had an acute subdural hemorrhage associated with bilateral transverse sinus occlusions. Split treatment dose LMWH was given, maintaining platelets >70 000/μL for 3 months. One year later, she had seizures associated with spontaneous acute on chronic subdural hemorrhage, new sagittal sinus thrombosis, and acute frontal intraparenchymal hemorrhage (platelet count, 54 000/μL). During a complicated 3-month admission, the patient developed a spontaneous left cerebellar hematoma with mild mass effect (prior platelet nadir, 47 000/μL). LMWH dosing for secondary thromboprophylaxis based on platelet counts were: platelets >70 000/μL, split standard treatment dose; platelets 50 000 to 70 000/μL, 75% split treatment dose; and platelets 30 000 to 50 000/μL, prophylactic dose. She gradually improved and returned to work.
Comments on case 2
This case illustrates the management of anticoagulant-refractory microvascular thrombosis and skin ulcers occurring with standard-intensity warfarin. The prevalence of the latter was 5.5% in one prospective study2 and 2% to 4%, depending on aPL phenotype, in another study.69 This patient’s course also highlights the challenge of anticoagulation management in the presence of recurrent intracranial hemorrhage associated with cerebral venous sinus thrombosis and thrombocytopenia. APS in patients with SLE is associated with a more complicated course and increased organ damage.70,71 Thrombocytopenia is associated with a two- to fourfold increased risk of thrombosis in patients with APS.72 The pathophysiology of thrombocytopenia in APS is not clear; probable mechanisms include autoantibodies against platelet glycoproteins as in immune thrombocytopenia, aPL-mediated platelet activation and consumption, and thrombotic microangiopathy. Our patient did not fulfill the preliminary classification criteria for definite or probable CAPS.73,74 Management was pragmatic, with IVMP, PEX, IVIG, and rituximab. The first 3 modalities are recommended by the McMaster RARE-Bestpractices guideline for CAPS.74 Rituximab, an anti-CD20 chimeric monoclonal antibody, is suggested for refractory cases. In the CAPS registry, 15 of 20 rituximab-treated patients survived.75 In the RITAPS study, 5 of 19 patients had skin ulcers, with complete and partial remission in 3 of 5 and 1 of 5 patients, respectively.68
In patients with anticoagulant-refractory thrombotic APS who have thrombocytopenia, LMWH is preferable to warfarin because of its shorter half-life, 3 to 6 hours,76 compared with 36 to 42 hours for warfarin.77 LMWH dosing is largely extrapolated from the cancer literature. However, high-quality evidence is lacking to inform guidelines, and recommendations are mainly based on expert opinion. After acute VTE in cancer patients, national and international guidance advises full-dose anticoagulation in patients with a platelet count >50 000/μL.78-81 The International Society on Thrombosis and Haemostasis suggests, in patients with thrombocytopenia (<50 000/μL) and a high risk of thrombus progression, platelet transfusion support to maintain platelet counts of ≥40 000 to 50 000/μL.78
The balance of thrombotic and hemorrhagic risk in critical sites is challenging and increased by thrombocytopenia. The recurrent intracerebral bleeds led us to set a higher platelet threshold for therapeutic anticoagulation for subsequent acute VTE. For secondary thromboprophylaxis, we reduced the dose of LMWH, based on the level of thrombocytopenia (as detailed earlier).
In this particular patient, IVIG and eltrombopag (a thrombopoietin agonist) were helpful. These therapies are widely used in the treatment of immune thrombocytopenia82 ; however, case reports/small series suggest that both may be associated with thrombosis in patients with APS and/or autoimmune rheumatic diseases.83-86 Notably, a review of 35 studies suggested that IVIG could be useful, in addition to standard therapy, to prevent recurrent thrombosis in patients with anticoagulant-refractory APS.87 The addition of antiplatelet treatment, a suggested option in patients with APS who rethrombose on therapeutic-intensity VKA,12,13,24 was precluded by thrombocytopenia. Iloprost, a prostacyclin analogue, was used intermittently in an attempt to improve the peripheral circulation.88,89 Its potential benefit in APS may go beyond vasodilation, possibly mediated by platelet function inhibition and endothelium-stabilizing properties. Epidermal grafting for wound healing involves the transfer of the epidermis from a healthy location to cover a wound and is a promising alternative to the more invasive conventional surgical techniques.90
We used eculizumab in this patient. Eculizumab is a humanized monoclonal antibody that binds complement protein C5 and prevents activation of the membrane attack complex, which leads to tissue injury. Case series suggest that it is beneficial for patients with SLE and/or APS with thrombotic microangiopathy.91 A phase 2a study of a C5a inhibitor, ALXN1007, was initiated in persistently aPL-positive patients with noncriteria APS manifestations, including thrombocytopenia, nephropathy, and/or skin ulcers. This study was terminated early after 9 patients were recruited, due to slow enrollment.92 Complement inhibition may be beneficial in APS patients with refractory microvascular thrombotic states, including thrombotic microangiopathy or, as in this case, chronic persistent microvascular thrombosis. A study in patients with thrombotic APS showed patient-derived aβ2GPI-induced complement activation in vitro, as indicated by a functional modified Ham assay, and increased C5b-9 deposition on the cell surface. These observations suggest that complement inhibition might be useful in patients with refractory thrombotic APS.93
There are no published studies on HBOT, also used in this patient, in APS. The largest RCT, in diabetic foot ulcers, reported a 26% (95% confidence interval, 10-38) improvement in amputation-free survival.94 However, because many patients did not complete treatment, the overall intention-to-treat analysis showed no benefit. HBOT may be an option for refractory APS-related ischemic cutaneous ulceration or tissue loss.
Case 3: anticoagulant-refractory digital ischemia with concomitant active SLE
A 22-year-old woman was diagnosed with SLE manifested by a skin rash, fever, serositis, and arthritis. Serologically, she had antibodies to DNA, Ro, Sm, and RNP. She developed bilateral avascular necrosis, requiring total hip replacements, and hypothyroidism. Her principal therapies included prednisolone, hydroxychloroquine, methotrexate, and MMF. Having been negative for all 3 aPL, she had a stroke at 41 years of age and was found to have isolated persistent high-titer IgG aß2GPI antibodies at >100 (NR, 0-10) U/mL. She commenced treatment on high-intensity warfarin, with a target INR of 3.5. At age 46 years, she developed finger ischemia and severe alopecia. Her platelet count, previously normal, dipped to 51 × 109/L. Although anti–double-stranded DNA antibody levels were normal, her complement C3 was low (0.49 g/L). The warfarin was switched to LMWH and iloprost added after platelet recovery, with the LMWH dose reduced to 75%. She also received IVMP, followed by oral prednisolone and rituximab, with subsequent MMF. The digital ischemia was persistent and painful. Digital sympathectomy did not prevent subsequent autoamputation of the first fingertip (Figure 4).
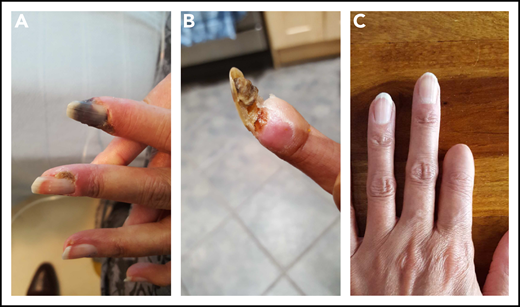
Photographs of disease progression in the left hand of patient 3. The early (A), middle (B), and end (C) phase of this patient’s digital gangrene affecting the index finger.
Photographs of disease progression in the left hand of patient 3. The early (A), middle (B), and end (C) phase of this patient’s digital gangrene affecting the index finger.
Comments on case 3
This case highlights the challenge of managing 2 diseases with different etiopathogenesis. SLE patients with concomitant APS present a major challenge. Thirty to forty percent of patients with SLE have aPL,95 but fewer develop relevant clinical features.70,96 This patient’s history highlights 2 management issues. First, there is uncertainty about optimal VKA intensity for APS-related stroke, which is reflected in the variation in guidelines.10-13 Second, how often should aPL be retested in patients with lupus who were initially aPL negative? When this patient first presented, she was aPL negative, but at age 41 years (20 years after her diagnosis), she had a stroke accompanied by high-titer isolated IgG aß2GPI. A further question arises as to how she might have been managed differently had aPL been detected during the 2-decade hiatus. Careful risk stratification, including assessment of aPL profile, cardiovascular risk prediction, and optimal management of concomitant autoimmune disease are important.97 The protective effect of low-dose aspirin for primary prevention of thrombosis in SLE patients with aPL is not supported by RCT data. Suggested approaches are consideration of low-dose aspirin in all non-thrombotic SLE patients with aPL12 or on a case-by-case basis.97
This patient had a digital sympathectomy to improve vasodilation. In this procedure, all neural connections between the digital nerve and artery are divided and the adventitia stripped from the main digital artery. This procedure may improve blood flow by interrupting sympathetic vasoconstrictor supply to the digital arteries and removing the external constrictive cuff or peri-adventitial fibrosis from around them.98 Although no formal studies have been undertaken, we have noted partial improvement in several patients with APS. This case illustrates that even single aPL-positive patients may develop major thrombotic manifestations.
Conclusions
Managing patients with anticoagulant-refractory thrombotic APS is a major challenge. However, with attention to detail, morbidity related to complex and severe thrombotic situations can be contained. Even the second patient, the most severely affected, returned to full-time work. The extent of the anticoagulant-refractory nature of patients with APS is highly variable. Thus, some patients can be managed successfully with increased VKA anticoagulation intensity. Others will require LMWH or fondaparinux and consideration of adding antiplatelet therapy. Further modalities include adjunctive treatment with hydroxychloroquine, statins, and vitamin D, as well as immunomodulation and complement inhibition; vascular options include vasodilators, epidermal grafting, digital sympathectomy, and HBOT. When anticoagulant-refractory patients have thrombocytopenia, balancing the risk of recurrent thrombosis vs bleeding becomes critical, with dose titration of anticoagulation based on platelet counts. Multicenter studies are required to guide the sequence of interventions and their comparative efficacy in patients with anticoagulant-refractory thrombotic APS.
Acknowledgment
D.A.I. acknowledges the support of the Biomedical Research Centre grant awarded to University College London and University College London Hospitals NHS Foundation Trust.
Authorship
Contribution: H.C. and D.A.I. contributed equally to the concept, design, first draft of the manuscript, and revision of the manuscript.
Conflict-of-interest disclosure: H.C. reports, outside the submitted work, institutional research support and support to attend scientific meetings from Bayer Healthcare, with honoraria for lectures from Bayer Healthcare and consultancy fees from UCB Biopharma paid to University College London Hospitals Charity. D.A.I. declares no competing financial interests.
Correspondence: Hannah Cohen, Haemostasis Research Unit, Department of Haematology, University College London, 1st Floor, 51 Chenies Mews, London WC1E 6HX, United Kingdom; e-mail: hannah.cohen@ucl.ac.uk.
