

Key Points
NETs promote fibrotic thrombus remodeling by TGF-β upregulation in thrombus fibroblasts.
Blocking NETs with DNase abrogates fibrotic remodeling.

Abstract
Acute pulmonary embolism generally resolves within 6 months. However, if the thrombus is infected, venous thrombi transform into fibrotic vascular obstructions leading to chronic deep vein thrombosis and/or chronic thromboembolic pulmonary hypertension (CTEPH), but precise mechanisms remain unclear. Neutrophils are crucial in sequestering pathogens; therefore, we investigated the role of neutrophil extracellular traps (NETs) in chronic thrombosis. Because chronic pulmonary thrombotic obstructions are biologically identical to chronic deep venous thrombi, the murine inferior vena cava ligation model was used to study the transformation of acute to chronic thrombus. Mice with staphylococcal infection presented with larger thrombi containing more neutrophils and NETs but less resolution. Targeting NETs with DNase1 diminished fibrosis and promoted thrombus resolution. For translational studies in humans, we focused on patients with CTEPH, a severe type of deep venous and pulmonary artery fibrotic obstruction after thrombosis. Neutrophils, markers of neutrophil activation, and NET formation were increased in CTEPH patients. NETs promoted the differentiation of monocytes to activated fibroblasts with the same cellular phenotype as fibroblasts from CTEPH vascular occlusions. RNA sequencing of fibroblasts isolated from thrombo-endarterectomy specimens and pulmonary artery biopsies revealed transforming growth factor-β (TGF-β) as the central regulator, a phenotype which was replicated in mice with fibroblast-specific TGF-β overactivity. Our findings uncover a role of neutrophil-mediated inflammation to enhance TGF-β signaling, which leads to fibrotic thrombus remodeling. Targeting thrombus NETs with DNases may serve as a new therapeutic concept to treat thrombosis and prevent its sequelae.
Introduction
Venous thromboembolism, which comprises deep vein thrombosis (DVT) and pulmonary embolism (PE), is a common and potentially fatal disease with an estimated incidence between 0.7 and 2 per 1000 individuals per year.1 Although venous thromboembolism is an acute disease, it may purport long-term complications. Postthrombotic syndrome is a frequent outcome of DVT, and chronic thromboembolic pulmonary hypertension (CTEPH) occurs as a late sequela following ≥1 PE.2
CTEPH, the model condition for nonresolving venous thrombosis, is a progressive pulmonary vascular disease estimated to occur in 0.4% to 6.2% of patients with a history of acute PE3 and also in patients without symptomatic PE.4 Two-thirds of all patients have confirmed previous DVT or both previous DVT and acute PE.5 Occlusive material in the lungs and in the deep veins are histologically identical.6 Histologic examination of thromboembolic material reveals red and white thrombi, which consist of myofibroblasts embedded in a collagen matrix and sparse inflammatory cells and areas of small vessels.7,8 Pulmonary endarterectomy (PEA) removing those vascular obstruction from the lungs is the treatment of choice for CTEPH.9 Previous studies from our group showed that infected pacemakers, ventriculo-atrial shunts for the treatment of hydrocephalus, and chronic inflammatory disorders such as osteomyelitis and inflammatory bowel disease, were associated with an increased risk of CTEPH.10 In line with this, we have demonstrated the role of staphylococcal infection in thrombus nonresolution using the vena cava ligation model and have provided support for this concept by clinical case correlations.11 Inflammatory cells are required for normal thrombus resolution because they promote both fibrinolysis and collagenolysis12,13 ; however, they could also play a role in misguided resolution. For example, as an innate reaction to inflammatory stimuli, neutrophils produce neutrophil extracellular traps (NETs), which are extracellular fibers primarily composed of DNA and histones that carry myeloperoxidase (MPO) and neutrophil elastase.14 NETs exert numerous biological responses such as antibacterial effects,14 cytotoxicity,15 pro-inflammatory cytokine release,16 and thrombin generation.17 The role of NETs in arterial and venous thrombosis18-21 and organ fibrosis22,23 has recently been reported. In our study, we are testing the hypothesis that NETs play a role in fibrotic transformation of fresh thrombus. Because clinically Staphylococcus aureus infection plays a role in CTEPH, we studied thrombosis in a murine infection model.
Methods
Patients and healthy controls
All procedures were approved by the Ethics Committee, Medical University of Vienna, Vienna, Austria (EK 220/2008). This study included 141 stable CTEPH patients at the Vienna General Hospital, and written informed consent was obtained from all patients and control participants. Hemodynamic data were collected at the time of baseline right heart catheterization. Patients were receiving vitamin K antagonists at the time of blood sampling. Sixty control participants were recruited in the Health and Prevention Center, Sanatorium Hera, Vienna, Austria (EK 1947/2014); they had a mean age of 61 years, and 35% were males. In this cohort, 13% of the participants reported a history of hypertension, 40% had dyslipidemia, 37% were smokers, and 5% were diabetic. No participant had ever experienced a coronary syndrome or venous thrombosis. Blood samples were centrifuged at 2000g for 10 minutes, and plasma was stored at −80°C. CTEPH tissue specimens acquired during PEA were fixed in 7.5% neutral buffered formalin and embedded in paraffin for subsequent histologic analysis.
Analysis of extracellular double-stranded DNA and citrullinated histone H3
Single radial enzyme diffusion and NET quantification assays
Total DNase activity was detected using a single radial enzyme diffusion assay as described27 with some modifications. Detailed protocol is provided in the supplemental Methods. For NET quantification assay, neutrophils (1 × 105) were seeded in 96-well plates at a concentration of 1 × 106 cells per mL and allowed to settle for 20 minutes before stimulation. S aureus was grown to stationary phase for 14 hours and centrifuged at 14 000g; the pellet was then resuspended in saline. Bacteria were added to neutrophils at different ratios of neutrophils to bacteria (1:5, 1:10, and 1:20) and incubated at 37°C for 60 minutes.28 The positive control for cell lysis was 10% Triton X-100. Sytox Green (50 µM) was added, and the plate was read at 450 nm using a Synergy H1 Hybrid Reader (BioTek). Results were computed relative to the positive control.
Monocyte isolation and stimulation
Monocytes were isolated from CTEPH patients using a Monocyte Enrichment Kit (StemCell), according to the manufacturer’s protocol. For stimulation experiments, monocytes (1 × 106 cells per mL) were seeded into a 12-well culture plate and allowed to adhere for 2 hours at 37°C. Nonadherent cells were carefully washed, and the adherent cells were stimulated for 7 days with isolated NETs (500 ng/mL), NETs with 40 IU/mL DNase1 (Dornase alfa, Pulmozyme), dsDNA (250 ng/mL), or vehicle control. At the end of the experiment, cells were prepared for flow cytometry, immunocytochemistry, and RNA isolation. Detailed protocols are provided in the supplemental Methods.
Fibroblast isolation from PEA specimens
Thrombus fibroblasts were isolated from thrombus material excised during PEA, and adventitial fibroblasts were isolated from a small pulmonary artery biopsy at the end of the endarterectomy from the same patient before closing the pulmonary artery incision (n = 4). Detailed protocol is given in the supplemental Methods.
RNA sequencing
Cellular RNA was extracted from nearly confluent fibroblasts using Reliaprep RNA Miniprep System according to the manufacturer’s instructions. RNA quality was estimated from 28S and 18S ribosomal RNA peaks on a Bioanalyzer 2100 instrument using the RNA 6000 Nano Kit. Samples with RNA integration number of 8 or more were further processed for RNA sequencing (RNA-seq) (Microsynth). The libraries were sequenced on NextSeq 500/550 platform to obtain ∼100 million uniquely mapped reads. Normalization of the raw counts and differential gene expression analysis were carried out using the R software package DESeq2 (version 1.18.1). RNA-seq data are available from Gene Expression Omnibus (GEO; GSE149413).
Animal experiments
All animal experiments were conducted in accordance with the guidelines and procedures approved by the Institutional Animal Care Committee (Medical University of Vienna) and the Austrian Ministry of Science (414/2018 and 287/2018). To avoid aggressive behavior and hierarchal structures of experimental animals during group housing29-31 that would affect their hormonal balance, only female mice were used in this study. Mouse models of infection and fibroblast-specific transforming growth factor-β (TGF-β) overactivity (TBRIIΔk), inferior vena cava ligation methodology, in vivo ultrasound imaging, and other animal experiments are described in the supplemental Methods.
Statistics and additional methodologies
Normality of distribution was determined using Kolmogorov-Smirnov and Shapiro-Wilk tests. Comparison between 2 groups was performed by 2-tailed Student t test or nonparametric Mann-Whitney U test. For multigroup comparisons, 1-way analysis of variance with Tukey’s post hoc correction was used. In case of normal distribution, data are represented as mean ± standard error of the mean (SEM); otherwise, median and interquartile ranges are provided. Data were analyzed with SPSS 26.0 (IBM Corp) and GraphPad Prism 8. A P value of <.05 was considered statistically significant. The supplemental Methods contains descriptions of human neutrophils and NETs isolation, histology, and immunostaining, real-time quantitative polymerase chain reaction (qPCR), enzyme-linked immunosorbent assays, flow cytometry, and immunoblotting.
Results
NETs are the major upstream trigger of fibrosis of infected venous thrombi
We have previously shown in patients and in the mouse model that thrombus infection delays thrombus resolution and causes fibrosis. Because infection is a potent natural inducer of neutrophilia and NET formation, we first assessed the involvement of NETs (Figure 1A). Mice infected with S aureus exhibited higher thrombus volume with delayed resolution, as measured by high-frequency ultrasound (Figure 1B; supplemental Figure 1A). In contrast, thrombus volume was markedly reduced on days 7 and 14 in mice receiving DNase1 (Figure 1B; supplemental Figure 1A). In mice infected with S aureus, neutrophil counts were increased on day 3 with no difference at later time points (Figure 1C). To assess the formation of NETs after infection, we measured circulating levels of CitH3 and dsDNA. Infected mice demonstrated elevated CitH3 on day 3 after thrombus induction, which was reduced after the administration of DNase1 (Figure 1D). Circulating extracellular dsDNA in infected mice showed a significant increase on day 14 after ligation (Figure 1E). Application of DNase1 resulted in marked reduction of plasma dsDNA on day 14 after ligation (Figure 1E). As expected, DNase activity was elevated on days 3 and 7 in mice receiving DNase1 (Figure 1F). High DNase activity persisted until 1 week after DNase1 administration had been discontinued (Figure 1F).
![NETs are the major upstream trigger of fibrosis of infected venous thrombi. (A) Workflow for thrombus induction, thrombus infection with S aureus, and DNase1 treatment. (B) Thrombus volume as measured by high-frequency ultrasound (HFUS) in control, infected, and DNase1-treated mice (n = 5 mice per group). (C) Blood neutrophil counts, (D) CitH3, (E) extracellular dsDNA, and (F) DNase activity in mouse plasma at different time points post ligation (n = 6-8 mice per group). (G) Representative Ly6G stainings (scale bars, 200 µm) and (H) CitH3 stainings in day 3 thrombus of different groups (n = 3-5 mice per group). Left: lower magnification (scale bars, 200 µm); right: higher magnification (scale bars, 20 µm) of boxed area. Bar graph quantifications are shown right below the respective histochemistry panels. (I) Representative trichrome staining and collagen area relative to total thrombus area (scale bars, 200 µm). (J) qPCR analysis for genes associated with fibrosis (n = 4-6 mice per group) in mouse thrombus at different time points after ligation. Data are represented as mean ± SEM. *P < .05; **P < .01; ***P < .001 (1-way analysis of variance [ANOVA] with Tukey’s post hoc correction). DAPI, 4′,6-diamidino-2-phenylindole; NE, neutrophil elastase.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/8/10.1182_blood.2020005861/1/m_bloodbld2020005861f1.png?Expires=1769129302&Signature=DdrboElWPIC~Me6OeDjCUauo-UQa7tji9vhqlKdMsIjI3GwlF-THiAFD5kBuO7W1PXm~4UZsF6~c4BNPeny1DwoUwRFqqk7AR6wMTBPuOrOiODGvtPyusMolnbptnV9D4KKcXHJFU54cdh3mEyIYuxmTDNhlAmSgZJ2JEv3wLUsDc57Ihc0LH95hrWD9cZaDRL9gFYXY-hkk2OZO9r9qRL78JG7NSDhbWkOglrgopb6rHUdvAyy9WeFYPu5P4JaGK89iTibHb5ciaF5eXm9ASCPeTkCSD~UazDgBOFIrH9OXQwlIwnPnRlr4OySlTwp5XQSqT8aMr0r2wY0E3OeUsg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
NETs are the major upstream trigger of fibrosis of infected venous thrombi. (A) Workflow for thrombus induction, thrombus infection with S aureus, and DNase1 treatment. (B) Thrombus volume as measured by high-frequency ultrasound (HFUS) in control, infected, and DNase1-treated mice (n = 5 mice per group). (C) Blood neutrophil counts, (D) CitH3, (E) extracellular dsDNA, and (F) DNase activity in mouse plasma at different time points post ligation (n = 6-8 mice per group). (G) Representative Ly6G stainings (scale bars, 200 µm) and (H) CitH3 stainings in day 3 thrombus of different groups (n = 3-5 mice per group). Left: lower magnification (scale bars, 200 µm); right: higher magnification (scale bars, 20 µm) of boxed area. Bar graph quantifications are shown right below the respective histochemistry panels. (I) Representative trichrome staining and collagen area relative to total thrombus area (scale bars, 200 µm). (J) qPCR analysis for genes associated with fibrosis (n = 4-6 mice per group) in mouse thrombus at different time points after ligation. Data are represented as mean ± SEM. *P < .05; **P < .01; ***P < .001 (1-way analysis of variance [ANOVA] with Tukey’s post hoc correction). DAPI, 4′,6-diamidino-2-phenylindole; NE, neutrophil elastase.
NETs are the major upstream trigger of fibrosis of infected venous thrombi. (A) Workflow for thrombus induction, thrombus infection with S aureus, and DNase1 treatment. (B) Thrombus volume as measured by high-frequency ultrasound (HFUS) in control, infected, and DNase1-treated mice (n = 5 mice per group). (C) Blood neutrophil counts, (D) CitH3, (E) extracellular dsDNA, and (F) DNase activity in mouse plasma at different time points post ligation (n = 6-8 mice per group). (G) Representative Ly6G stainings (scale bars, 200 µm) and (H) CitH3 stainings in day 3 thrombus of different groups (n = 3-5 mice per group). Left: lower magnification (scale bars, 200 µm); right: higher magnification (scale bars, 20 µm) of boxed area. Bar graph quantifications are shown right below the respective histochemistry panels. (I) Representative trichrome staining and collagen area relative to total thrombus area (scale bars, 200 µm). (J) qPCR analysis for genes associated with fibrosis (n = 4-6 mice per group) in mouse thrombus at different time points after ligation. Data are represented as mean ± SEM. *P < .05; **P < .01; ***P < .001 (1-way analysis of variance [ANOVA] with Tukey’s post hoc correction). DAPI, 4′,6-diamidino-2-phenylindole; NE, neutrophil elastase.
Thrombi of mice infected with S aureus had higher neutrophil counts as observed by Ly6G staining that did not change after DNase1 application (Figure 1G). In contrast, infected mice exhibited higher thrombus CitH3 positivity on day 3, which was reduced after treatment with DNase1 (Figure 1H). Trichrome staining of mouse thrombi at different time points over a period of 14 days illustrated more collagen in infected thrombi that was reduced after administration of DNase1 (Figure 1I). Simultaneous upregulation in genes associated with neutrophil inflammation (neutrophil elastase, matrix metalloproteinase-9 [MMP-9]) and fibrosis (TGFB1, Col3a1) were observed in thrombi from mice infected with S aureus (Figure 1J; supplemental Figure 1B). This effect was significantly reduced in thrombi from mice treated with DNase1 (Figure 1J; supplemental Figure 1B). These experiments underline a significant contribution of NETs in mediating fibrotic remodeling of infected mouse thrombi.
Neutrophil inflammation is common in CTEPH
To verify the presence of neutrophil inflammation and NETs in patients with chronic thrombosis, we measured circulating levels of neutrophils, neutrophil components, and NET markers in CTEPH patients and healthy participants. Characteristics of patients and controls are detailed in Table 1. Neutrophil counts (Figure 2A) and the neutrophil activation and degranulation marker MPO were significantly elevated in CTEPH (Figure 2B). Furthermore, plasma levels of MMP-9 antigen and the functionally active protease were high in CTEPH patients (Figure 2C-D). Concentrations of dsDNA were increased (Figure 2E), and plasma DNase activity was very low (Figure 2F). No differences were observed between patients and healthy participants in circulating levels of CitH3 (Figure 2G); however, CitH3 positivity was observed in fresh red fibrin-rich parts of CTEPH vascular occlusions (Figure 2H-I), indicating the presence of NETs.
Characteristics of patients and controls
| Characteristic | CTEPH patients (n = 141) | Controls (n = 60) |
|---|---|---|
| Mean age, y | 58.7 ± 13.5 | 61.0 ± 6.7 |
| Sex | ||
| Male | 58 | 21 |
| Female | 83 | 39 |
| Body-mass-index, kg/m2 | 26.6 (24.0-30.7) | 25.3 (22.6-27.9)* |
| WHO functional class | ||
| I | 5 | 60 |
| II | 23 | 0 |
| III | 84 | 0 |
| IV | 29 | 0 |
| Hemodynamic parameters | ||
| Pulmonary arterial pressure, mmHg | ||
| Systolic | 82.0 (63.0-101.7) | NA |
| Diastolic | 27.0 (20.0-35.0) | NA |
| Mean | 47.0 (35.0-55.7) | NA |
| Mean wedge | 9.0 (7.0-13.0) | NA |
| Pulmonary vascular resistance, dynes/s/cm−5 | 654 (333-894) | NA |
| Arterial saturation, % | 91.0 (86.0-93.2) | NA |
| Cardiac output, L/min | 4.6 (3.9-5.5) | NA |
| Cardiac index, L/min/m2 | 2.4 (2.1-2.8) | NA |
| Laboratory parameters | ||
| Leukocytes, G/L | 6.7 (5.7-8.3) | 5.8 (5.0-7.0)* |
| Neutrophils, G/L | 4.5 (3.5-5.3) | 3.4 (2.8-4.0)* |
| Fibrinogen, mg/dL | 360.0 (310.5-436.5) | NA |
| C-reactive protein, mg/dL | 0.3 (0.1-0.8) | 0.4 (0.1-1.0) |
| Serum amyloid A, mg/L | 8.3 (5.9-14.1) | NA |
| NT-proBNP, pg/mL | 1099.0 (230.4-2694.0) | NA |
| Lower extremity conditions | ||
| Previous DVT, lower leg trauma, surgeries, wound infections, or venous ulcers | 73 | 0 |
| Characteristic | CTEPH patients (n = 141) | Controls (n = 60) |
|---|---|---|
| Mean age, y | 58.7 ± 13.5 | 61.0 ± 6.7 |
| Sex | ||
| Male | 58 | 21 |
| Female | 83 | 39 |
| Body-mass-index, kg/m2 | 26.6 (24.0-30.7) | 25.3 (22.6-27.9)* |
| WHO functional class | ||
| I | 5 | 60 |
| II | 23 | 0 |
| III | 84 | 0 |
| IV | 29 | 0 |
| Hemodynamic parameters | ||
| Pulmonary arterial pressure, mmHg | ||
| Systolic | 82.0 (63.0-101.7) | NA |
| Diastolic | 27.0 (20.0-35.0) | NA |
| Mean | 47.0 (35.0-55.7) | NA |
| Mean wedge | 9.0 (7.0-13.0) | NA |
| Pulmonary vascular resistance, dynes/s/cm−5 | 654 (333-894) | NA |
| Arterial saturation, % | 91.0 (86.0-93.2) | NA |
| Cardiac output, L/min | 4.6 (3.9-5.5) | NA |
| Cardiac index, L/min/m2 | 2.4 (2.1-2.8) | NA |
| Laboratory parameters | ||
| Leukocytes, G/L | 6.7 (5.7-8.3) | 5.8 (5.0-7.0)* |
| Neutrophils, G/L | 4.5 (3.5-5.3) | 3.4 (2.8-4.0)* |
| Fibrinogen, mg/dL | 360.0 (310.5-436.5) | NA |
| C-reactive protein, mg/dL | 0.3 (0.1-0.8) | 0.4 (0.1-1.0) |
| Serum amyloid A, mg/L | 8.3 (5.9-14.1) | NA |
| NT-proBNP, pg/mL | 1099.0 (230.4-2694.0) | NA |
| Lower extremity conditions | ||
| Previous DVT, lower leg trauma, surgeries, wound infections, or venous ulcers | 73 | 0 |
Data are represented as no., mean ± standard deviation, or median (interquartile range).
NA, not available; NT-proBNP, N-terminal probrain natriuretic peptide; WHO, World Health Organization.
Descriptive P < .05 (derived from Mann-Whitney U test).
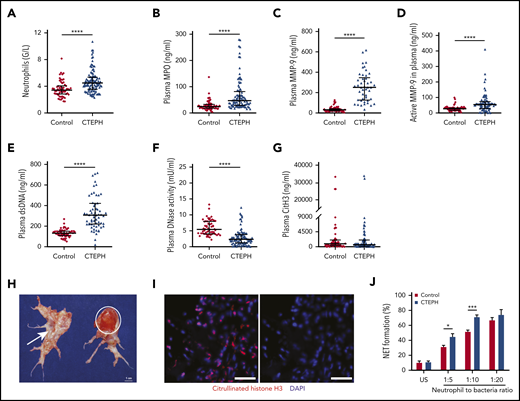
Neutrophil inflammation is common in CTEPH. (A) Circulating blood neutrophil counts: control (n = 60), CTEPH (n = 109); (B) plasma concentrations of MPO: control (n = 60), CTEPH (n = 96), (C) total MMP-9: control (n = 60), CTEPH (n = 46), (D) active MMP-9: control (n = 60), CTEPH (n = 67), (E) extracellular dsDNA: control (n = 60), CTEPH (n = 63), (F) DNase activity: control (n = 57), CTEPH (n = 103), and (G) circulating CitH3: control (n = 60), CTEPH (n = 77). (H) Macroscopic view of thrombus excised during pulmonary endarterectomy representing fresh (red; circle) and organized (white; arrow) material. (I) Immunofluorescence staining for CitH3 in fresh red CTEPH thrombus (scale bars, 50 µm). Data in panels A-G are shown as median and interquartile ranges. (J) Ex vivo NET formation in healthy controls (n = 5) and in patients with CTEPH (n = 6) after coincubation with S aureus. Data are shown as mean ± SEM. *P < .05; ***P < .001 (unpaired Student t test); ****P < .0001 (Mann-Whitney U test). US, unstimulated.
Neutrophil inflammation is common in CTEPH. (A) Circulating blood neutrophil counts: control (n = 60), CTEPH (n = 109); (B) plasma concentrations of MPO: control (n = 60), CTEPH (n = 96), (C) total MMP-9: control (n = 60), CTEPH (n = 46), (D) active MMP-9: control (n = 60), CTEPH (n = 67), (E) extracellular dsDNA: control (n = 60), CTEPH (n = 63), (F) DNase activity: control (n = 57), CTEPH (n = 103), and (G) circulating CitH3: control (n = 60), CTEPH (n = 77). (H) Macroscopic view of thrombus excised during pulmonary endarterectomy representing fresh (red; circle) and organized (white; arrow) material. (I) Immunofluorescence staining for CitH3 in fresh red CTEPH thrombus (scale bars, 50 µm). Data in panels A-G are shown as median and interquartile ranges. (J) Ex vivo NET formation in healthy controls (n = 5) and in patients with CTEPH (n = 6) after coincubation with S aureus. Data are shown as mean ± SEM. *P < .05; ***P < .001 (unpaired Student t test); ****P < .0001 (Mann-Whitney U test). US, unstimulated.
To understand NET formation (NETosis) in CTEPH, we stimulated neutrophils isolated from patients by coincubation ex vivo with S aureus. Neutrophils from CTEPH patients were more prone to undergo NET formation in the presence of S aureus than were neutrophils from healthy participants (Figure 2J), and this response was similar to the response when ionomycin was used to induce NETosis (data not shown). These findings confirm a chronic state of activation of CTEPH neutrophils.
NETs promote fibroblast differentiation
To understand how NETs are connected to the fibrotic response of chronic thrombosis, we investigated the effects of NETs and NET-associated components on fibroblast differentiation and activation in CTEPH patients. To this end, monocytes isolated from CTEPH patients (supplemental Figure 2) were coincubated with either dsDNA, isolated NETs, or NETs plus DNase.
In the presence of NETs, transdifferentiated cells expressed macrophage surface markers as computed by flow cytometry to account for 5% TLR4, 11% CD163, 18% CD206, and 13% fibroblast-specific protein–positive cells (Figure 3A; supplemental Figure 3). Differentiated cells exhibited mixed morphology (round and spindle-shaped), with positive expression of mesenchymal marker α smooth muscle actin (α-SMA) and fibroblast activation protein (FAP) (Figure 3B). qPCR analysis revealed significant upregulation of the TGF-β signaling pathway–associated genes such as TGFB1, SMAD3, Col1a1, and Col3a1 (Figure 3C). Together, these results indicate that NET-dependent monocyte differentiation leads to a predominantly fibroblast phenotype with upregulation of TGF-β–dependent signal transduction.
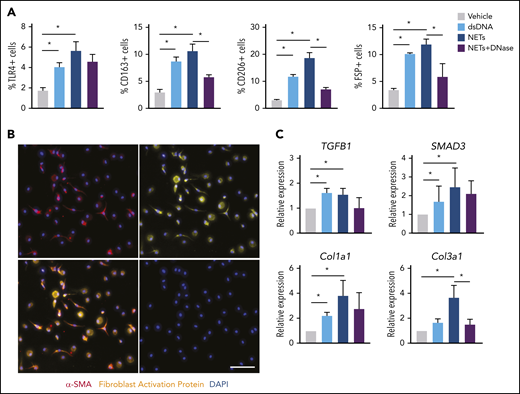
NETs promote fibroblast differentiation. (A) Characterization of differentiated cells after coincubation of CTEPH monocytes with dsDNA, isolated NETs, NETs plus DNase, or vehicle for their surface expression of TLR4, CD163, CD206, and fibroblast-specific protein (FSP) by flow cytometry (n = 4). The percentage of positive cells of single events was computed. (B) Representative coimmunofluorescent staining of α smooth muscle actin (α-SMA) (red) and fibroblast activation protein (FAP) (yellow) in CTEPH monocytes exposed to NETs (scale bars, 100 µm). (C) qPCR analysis for TGF-β signaling pathway genes in differentiated cells. Data are shown as mean ± SEM. *P < .05 (1-way ANOVA with Tukey’s post hoc correction).
NETs promote fibroblast differentiation. (A) Characterization of differentiated cells after coincubation of CTEPH monocytes with dsDNA, isolated NETs, NETs plus DNase, or vehicle for their surface expression of TLR4, CD163, CD206, and fibroblast-specific protein (FSP) by flow cytometry (n = 4). The percentage of positive cells of single events was computed. (B) Representative coimmunofluorescent staining of α smooth muscle actin (α-SMA) (red) and fibroblast activation protein (FAP) (yellow) in CTEPH monocytes exposed to NETs (scale bars, 100 µm). (C) qPCR analysis for TGF-β signaling pathway genes in differentiated cells. Data are shown as mean ± SEM. *P < .05 (1-way ANOVA with Tukey’s post hoc correction).
Thrombus fibrosis in CTEPH is linked with TGF-β
Macroscopically, CTEPH thrombus contains fresh frail fibrin-rich material (red) and organized collagen-rich tissues (white) (Figure 2H). Baseline expression of TGFB1 was significantly higher in white thrombus than in the pulmonary artery of the same patient (Figure 4A). To characterize the collagen composition of a white CTEPH thrombus, we evaluated the levels of 4 major collagen types—Col1a1, Col2a1, Col3a1, and Col4a1. qPCR and immunoblot of thrombus tissue revealed a high expression of Col1a1, followed by Col3a1 and Col4a1, whereas Col2a1 was low or not expressed (Figure 4B-C). At the cellular level, trichrome staining of white CTEPH thrombus demonstrated purple fibroblast-like cells embedded in a green collagen bed (Figure 4D). Coimmunostaining for α-SMA and FAP confirmed the presence of active fibroblasts in CTEPH thrombus (Figure 4E).
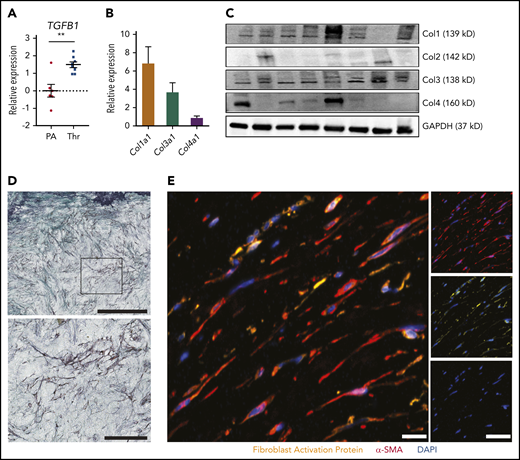
Fibrotic remodeling of CTEPH thrombi. (A) qPCR analysis for TGFB1 expression in CTEPH thrombus (Thr) (n = 8) and nonthrombosed pulmonary artery (PA) (n = 6). (B) qPCR analysis (n = 22) and (C) immunoblotting (n = 8) for different collagen types in CTEPH thrombus. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was chosen as a housekeeping protein. (D) Representative trichrome staining of CTEPH thrombus (scale bar, 200 µm) and magnification of the boxed area shows fibroblast-like cells (scale bar, 100 µm). (E) Merged immunofluorescence staining for α-SMA (red) and FAP (yellow) of CTEPH thrombus; single-channel images are shown on the right (scale bars, 20 µm). Data in panel A are shown as mean ± SEM. **P < .01 (unpaired Student t test).
Fibrotic remodeling of CTEPH thrombi. (A) qPCR analysis for TGFB1 expression in CTEPH thrombus (Thr) (n = 8) and nonthrombosed pulmonary artery (PA) (n = 6). (B) qPCR analysis (n = 22) and (C) immunoblotting (n = 8) for different collagen types in CTEPH thrombus. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was chosen as a housekeeping protein. (D) Representative trichrome staining of CTEPH thrombus (scale bar, 200 µm) and magnification of the boxed area shows fibroblast-like cells (scale bar, 100 µm). (E) Merged immunofluorescence staining for α-SMA (red) and FAP (yellow) of CTEPH thrombus; single-channel images are shown on the right (scale bars, 20 µm). Data in panel A are shown as mean ± SEM. **P < .01 (unpaired Student t test).
To obtain an unbiased and comprehensive understanding of genes dysregulated in thrombus fibroblasts, we used RNA-seq to study the transcriptome profile of fibroblasts isolated from pairs of CTEPH thrombus and pulmonary artery adventitia of the same patient (Figure 5A; supplemental Figure 4). Twenty-four genes were found to be differentially expressed in thrombus fibroblasts with fold change ≥2 (Padj < .05) vs adventitial fibroblasts (Figure 5B). qPCR analysis of 3 upregulated genes (MMP-9, PLA2G5, and CLDN14) in a larger patient cohort confirmed expression similar to that shown by RNA-seq (Figure 5C). Genes that showed significant changes were associated with the TGF-β pathway.32-35 Fibroblasts isolated from CTEPH thrombi demonstrated higher expression of TGFB1, Col1a1, and Col3a1 than adventitial fibroblasts (Figure 5D). These findings indicate that upregulation of TGF-β signaling is associated with the intravascular remodeling process.
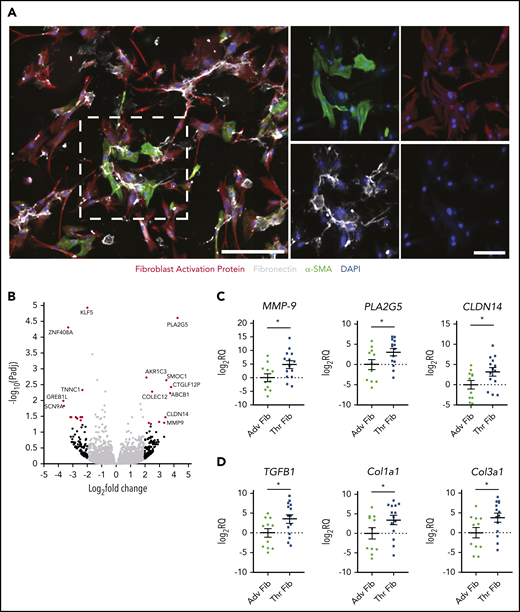
Transcriptome profiling of CTEPH thrombus fibroblasts. (A) Immunofluorescence staining of fibroblasts isolated from CTEPH thrombus for FAP (red), matrix protein fibronectin (white), and α-SMA (green), with DAPI as the nuclear stain. Left: lower magnification (scale bar, 100 µm); right: higher magnification (scale bars, 20 µm) of the boxed area with single-channel images. (B) Volcano plot illustrating fold differences in gene expression (genes with fold change ≥2 are highlighted in red, black dots represent genes with nonsignificant expression differences, and gray dots show genes with a fold change of <2) of thrombus fibroblasts and corresponding adventitial fibroblasts from CTEPH patients (n = 4). (C) qPCR validation of 3 upregulated genes and (D) TGF-β pathway–associated genes in a larger cohort (n = 11-14). Data are shown as mean ± SEM. Adv Fib, adventitial fibroblasts; Thr Fib, thrombus fibroblasts; CLDN14, Claudin 14; PLA2G5, phospholipase A2 Group V. *P < .05 (unpaired Student t test).
Transcriptome profiling of CTEPH thrombus fibroblasts. (A) Immunofluorescence staining of fibroblasts isolated from CTEPH thrombus for FAP (red), matrix protein fibronectin (white), and α-SMA (green), with DAPI as the nuclear stain. Left: lower magnification (scale bar, 100 µm); right: higher magnification (scale bars, 20 µm) of the boxed area with single-channel images. (B) Volcano plot illustrating fold differences in gene expression (genes with fold change ≥2 are highlighted in red, black dots represent genes with nonsignificant expression differences, and gray dots show genes with a fold change of <2) of thrombus fibroblasts and corresponding adventitial fibroblasts from CTEPH patients (n = 4). (C) qPCR validation of 3 upregulated genes and (D) TGF-β pathway–associated genes in a larger cohort (n = 11-14). Data are shown as mean ± SEM. Adv Fib, adventitial fibroblasts; Thr Fib, thrombus fibroblasts; CLDN14, Claudin 14; PLA2G5, phospholipase A2 Group V. *P < .05 (unpaired Student t test).
NET-induced thrombus fibrosis is dependent on TGF-β
To confirm the role of TGF-β in thrombus resolution, we used transgenic mice with TGF-β overactivity in fibroblasts (TBRIIΔk) in the stagnant flow venous thrombosis model (Figure 6A-B). Thrombus formation and resolution were monitored by high-frequency ultrasound (as represented in Figure 6C). Thrombus lengths, volumes, and weights were higher on days 7, 14, and 21 in TBRIIΔk mice than in controls (Figure 6D-F). To better understand the phenotype of TBRIIΔk mice, we measured tail bleeding time, activated platelets, platelet-leukocyte and -neutrophil aggregates, and thrombin generation time in control and TBRIIΔk mice at baseline. No differences were observed in both groups other than nonsignificantly higher platelet-leukocyte aggregates in TBRIIΔk mice (supplemental Figures 5 and 6). Because TBRIIΔk mice showed enhanced primary thrombus formation, we measured the plasma levels of active TGF-β1, D-dimer, plasminogen activator inhibitor-1 (PAI-1), coagulation factor XII (FXII), and procoagulant platelet factor 4 (PF4). Active TGF-β1 and D-dimer concentrations were increased in TBRIIΔk mice at baseline and on day 14 (supplemental Figure 7A-B). Levels of PAI-1 and FXII were increased on days 7 and 14 in TBRIIΔk mice; however, no difference was observed in plasma levels of PF4 (supplemental Figure 7C-E). Thrombi from TBRIIΔk mice demonstrated more collagen-rich areas on days 14 and 21 (Figure 6G) and an upregulation of CD68, MMP-2, MMP-9, and genes involved in fibrosis (Figure 6H).
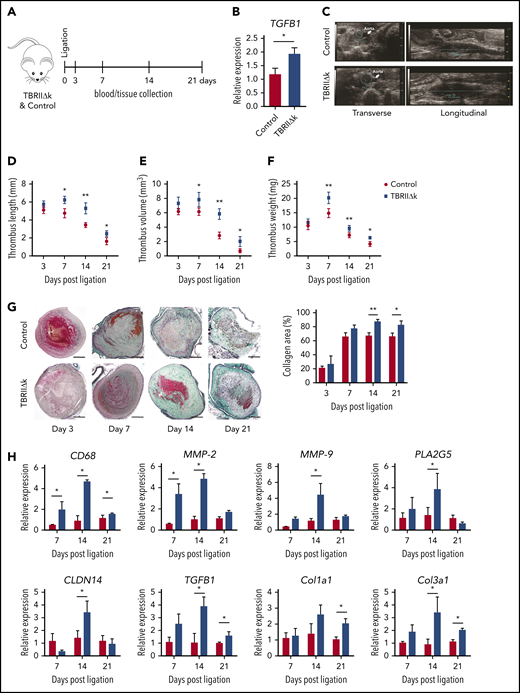
Thrombus resolution in mice with fibroblast-specific TGF-β overactivity. (A) Workflow for thrombus induction and (B) qPCR analysis for TGFB1 expression in lungs of control and TBRIIΔk mice at baseline (n = 4 mice per group). (C) Representative HFUS images showing thrombus area (left) and length (right) measurement on day 7 after ligation in control and TBRIIΔk mice. (D) Thrombus length, (E) thrombus volume as measured by HFUS (n = 5 mice per group), and (F) harvested thrombus weight (n = 5-8 mice per group). (G) Representative trichrome staining of mouse thrombus at different time points after ligation and collagen area represented relative to total thrombus area (n = 5-7 mice per group). Right: bar graph quantification for both groups is shown next to histochemistry panels (scale bars, 200 µm). (H) qPCR analysis of mouse thrombus at different time points (n = 4-6 mice per group). Data are shown as mean ± SEM. *P < .05; **P < .01 (unpaired Student t test).
Thrombus resolution in mice with fibroblast-specific TGF-β overactivity. (A) Workflow for thrombus induction and (B) qPCR analysis for TGFB1 expression in lungs of control and TBRIIΔk mice at baseline (n = 4 mice per group). (C) Representative HFUS images showing thrombus area (left) and length (right) measurement on day 7 after ligation in control and TBRIIΔk mice. (D) Thrombus length, (E) thrombus volume as measured by HFUS (n = 5 mice per group), and (F) harvested thrombus weight (n = 5-8 mice per group). (G) Representative trichrome staining of mouse thrombus at different time points after ligation and collagen area represented relative to total thrombus area (n = 5-7 mice per group). Right: bar graph quantification for both groups is shown next to histochemistry panels (scale bars, 200 µm). (H) qPCR analysis of mouse thrombus at different time points (n = 4-6 mice per group). Data are shown as mean ± SEM. *P < .05; **P < .01 (unpaired Student t test).
We also examined whether NET-induced thrombus fibrosis (as seen in Figure 1) is TGF-β dependent. To this end, we used S aureus infection in control and TBRIIΔk mice (Figure 7A) and observed elevated NET markers in the transgenic mice (data not shown). Plasma levels of active TGF-β1 (Figure 7B) and thrombus weights (Figure 7C) were higher in TBRIIΔk mice with infection on day 21 after ligation. More collagen-rich area (Figure 7D) and increased pro-fibrotic gene expression (Figure 7E) were observed in thrombus from infected TBRIIΔk mice 21 days after ligation. Together, these results provide compelling evidence that the connection between NET functions and TGF-β signaling can drive thrombus persistence and fibrosis.

Infection and TGF-β overactivity aggravate thrombus fibrosis. (A) Workflow for thrombus induction and infection. (B) Plasma levels of active TGF-β1 and (C) harvested thrombus weights on day 21 after ligation (n = 5-6 mice per group). (D) Representative trichrome staining of day 21 mouse thrombus and collagen area relative to total thrombus area (n = 4-5 mice per group) (scale bars, 200 µm). (E) qPCR analysis of profibrotic genes in mouse thrombus on day 21 after ligation (n = 5-6 mice per group). Data are shown as mean ± SEM. *P < .05 (unpaired Student t test).
Infection and TGF-β overactivity aggravate thrombus fibrosis. (A) Workflow for thrombus induction and infection. (B) Plasma levels of active TGF-β1 and (C) harvested thrombus weights on day 21 after ligation (n = 5-6 mice per group). (D) Representative trichrome staining of day 21 mouse thrombus and collagen area relative to total thrombus area (n = 4-5 mice per group) (scale bars, 200 µm). (E) qPCR analysis of profibrotic genes in mouse thrombus on day 21 after ligation (n = 5-6 mice per group). Data are shown as mean ± SEM. *P < .05 (unpaired Student t test).
Discussion
Fibrosis is a hallmark of chronic nonresolving thrombus, but mechanisms leading to fibrotic vascular obstructions remain elusive. In this study, we identified a role of neutrophil inflammation and NETs in fibrotic remodeling of vascular thrombus. To translate these findings to human medicine, we have performed studies in CTEPH, a condition with chronic pulmonary arterial obstructions. Our data suggest that NETs are the upstream trigger of TGF-β–mediated thrombus fibrosis. Clinically, venous thrombosis in the context of neutrophil inflammation is prone to transform into chronic thrombosis.
Neutrophils are primary responders to an acute inflammatory stimuli or invading pathogens. They promote bacterial clearance through phagocytosis, production of reactive oxygen species, release of pro-inflammatory cytokines, and liberation of decondensed chromatin as dense bactericidal DNA-histone webs called NETs. Apart from their classical role in mediating an early immune response, neutrophils are also relevant in propagating inflammation and thrombosis. In patients with acute PE, the neutrophil:lymphocyte ratio is increased36 and is an independent predictor of mortality.37 Further evidence for the association between neutrophilic inflammation and thrombosis, specifically in CTEPH, comes from the observation of higher neutrophil counts; elevated biomarkers for neutrophil activation; an association with DVT; lower leg trauma, surgeries, wound infections; or venous ulcers that were confirmed in 50% of patients in this study. There are other noninfectious conditions associated with neutrophilia such as familial Mediterranean fever, a hereditary inflammatory disorder with massive NET formation in the acute phase of the disease38 (1 patient in our cohort), and Behcets disease, a multisystem inflammatory disorder with periodic predominant neutrophilic inflammation leading to CTEPH.39,40 Despite this clinical evidence, we found low CitH3 plasma levels in CTEPH patients, presumably because neutrophil inflammation occurs in an episodic pattern. Alternative mechanisms involving NADPH (reduced form of nicotinamide adenine dinucleotide phosphate) oxidase41 could also account for low levels of CitH3 in these patients.
NETs are essential for pathogen clearance, but individual NET components such as cell-free DNA (cfDNA), nucleosomes, and MPO may enter the circulation to induce systemic hypercoagulability and thrombosis.42,43 Negatively charged cfDNA can activate the intrinsic pathway of coagulation via auto-activation of FXI and FXII, whereas NET-associated enzymes such as elastase and cathepsin G can promote extrinsic pathway activation by degrading tissue factor pathway inhibitor.44 Therefore, neutrophil inflammation may promote acute thrombosis directly, as was observed in infected mice.
After a thromboembolic event, CTEPH evolves over months and years, and CTEPH thrombus specimens represent end-stage material of a vascular remodeling process originating from acute thrombus that transforms into a nonresolving fibrotic vascular obstruction. We observed that thrombus material is enriched in activated fibroblasts and myofibroblast-like cells causing excessive collagen production. Recently, it was shown that NETs and neutrophil interleukin-17 promoted differentiation and function of lung fibroblasts.45
Here we report that circulating neutrophils from CTEPH patients are activated, and NETs are able to transdifferentiate CTEPH monocytes ex vivo to fibroblast-like cells. Access to adventitial fibroblasts from the same patient provided a unique opportunity to study the transcriptome of thrombus-associated fibroblasts by RNA-seq. These cells from patients exhibited a TGF-β signature similar to that of in vitro transformed cells. In vivo, fibroblast-specific TGF-β overactivity in mice led to increased thrombus burden and delayed resolution with concomitant upregulation of collagens as observed in human white CTEPH thrombi. The presence of NETs and TGF-β overactivity aggravated the fibrotic response in murine venous thrombosis. These results underscore the role of TGF-β in NET-induced thrombus fibrosis.
The degradation of NETs by DNases is a natural counter-regulatory mechanism to balance excessive NET formation and action.14 Two predominant DNases have been identified: DNase1 and DNase 1–like 3 (DNase1L3) that independently degrade NETs. DNase1 is expressed by nonhematopoietic tissues and cleaves naked DNA, whereas DNase1L3 is secreted by immune cells and targets DNA-protein complexes.27 Although origin and substrate affinity differ, DNase1 and DNase1L3 were found to provide dual protection against detrimental effects of intravascular NETs.27 Previous reports have documented that administration of DNase1 protected mice from DVT.18,46 In our work, the administration of DNase1 diminished thrombus fibrosis. When administered 1 day after thrombus induction, DNase1 had a predominant effect on clearance of local NETs. It is plausible that the increase in DNase activity 1 week after termination of DNase administration is a result of endogenous DNase activity. Because dysfunctional coagulation is a prominent feature of bacterial infection, the potential effect of DNase1 on coagulation and an additional role of DNase1L3 cannot be ruled out.47
In conclusion, we have uncovered a role of neutrophil inflammation and NETs in chronic thrombosis and have demonstrated that NETs and their components mediate fibrotic remodeling of thrombi by augmenting TGF-β signaling. By using 2 mouse models, we have documented the roles of NETs and TGF-β in thrombus persistence. These new results provide a mechanistic explanation for NET-mediated fibrotic vascular remodeling in CTEPH. Targeting NETs with DNases in the acute stage of inflammatory thrombosis may prevent thrombus persistence.
Our study has strengths, but also limitations. CTEPH is clinically associated with S aureus infection,11 and therefore, we used the inferior vena cava ligation/infection model to replicate human disease. NETosis is a well-orchestrated sequence of cellular events in neutrophils upon activation, including disassembly of the cellular cytoskeleton, fragmentation of the endomembrane, rounding of the nucleus, permeabilization of the plasma membrane, and finally rupture of the nuclear and plasma membrane.48 The underlying cellular mechanisms of NETosis are still poorly defined. An NADPH oxidase-dependent pathway has been described that can be simulated by phorbol 12-myristate 13-acetate49 and a peptidylarginine deiminase 4 (PAD4)–dependent pathway that is driven by posttranslational histone citrullination. However, in the granulocyte colony-stimulating factor–induced neutrophilia model, NETosis occurs in the absence of PAD4,24 and a separate unique, very rapid oxidant-independent mechanism is characteristic of S aureus–induced NET formation28 that withstands a blockade of PAD4-mediated NET formation.50,51 On the basis of these evidences, we did not use the PAD4 knockout mice in the infection model; instead, we used DNase1 to degrade NETs.
One main shortfall of the model is that we draw conclusions on chronic thrombosis using a 21-day mouse model of stagnant flow venous thrombosis. Although this model resembles the time course and clinical features of human DVT,7,46,52 it might not completely replicate pulmonary embolism and chronic thrombosis in humans. However, CTEPH pulmonary arteries show the same pathophysiologic changes as with DVT,6,53 and the vena cava ligation model has been successfully used for CTEPH research.6,11,54,55 After nonocclusive vena cava ligation, thrombosis builds up over the first 3 days, overlapping with thrombus resolution that continues over the subsequent 30 days in the model.54 To account for the balance between thrombus formation and resolution, we have calculated relative volume changes as previously described54 in addition to ultrasound volumetry and direct histology. The infiltration of inflammatory neutrophils,52 monocytes, and endothelial cells is followed by retraction of the thrombus away from the vein wall, myofibroblast activation, and the formation of new vessels within and around the thrombus,7 with conserved patterns of RNA expression. Hemodynamic and laboratory parameters were not available from all CTEPH patients. The use of pulmonary artery adventitial tissues as the comparator for RNA-seq experiments may not be rated as normal tissues.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
The RNA-seq data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE149413).
Study data may be obtained by e-mail to the corresponding author.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Christopher Denton for providing breeding pairs of TBRIIΔk mice. The visual abstract was created in BioRender.
This work was supported by a research grant from the Austrian Science Fund (FWF SFB-F54; “Cellular Mediators Linking Inflammation and Thrombosis”).
Authorship
Contribution: S.S., and I.M.L. conceived and designed the study; S.S., T.M.H., A.S.O., S.C., A.A., T.A., J.R., A.P., I.S., A.M., R.W., E.W.-K., B.M., S.T., and W.K. acquired and analyzed the data; S.S., T.M.H., A.S.O., and I.M.L. interpreted and analyzed the data; and S.S., K.T.P., and I.M.L. drafted and critically revised the manuscript.
Conflict-of-interest disclosure: I.M.L. served as an investigator in trials, served as a consultant, received research grants, and was a member of scientific advisory boards for AOP Orphan Pharmaceuticals, Actelion, Merck Sharp & Dohme, Ferrer, Neutrolis, and United Therapeutics. The remaining authors declare no competing financial interests.
Correspondence: Irene M. Lang, Department of Internal Medicine II, Division of Cardiology, Medical University of Vienna, Währinger Guertel 18-20, 1090 Vienna, Austria; e-mail: irene.lang@meduniwien.ac.at.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal