

In this issue of Blood, report that activation of the MAPK pathway can compensate for loss of PI3K signaling in patients with CLL who did not respond to treatment with idelalisib.1
The use of kinase inhibitors in cancer holds great promise and has benefited patients with CLL with the approval of drugs that target the B-cell receptor signaling pathways, phosphoinositide 3-kinase δ (PI3Kδ; idelalisib) and BTK (ibrutinib). Idelalisib and ibrutinib target these kinases, leading to impressive response rates rarely seen in cancer.2,3 Inevitably, however, most cancers develop resistance to kinase inhibitors. The best understood resistance mechanisms are characterized by somatic mutations acquired by cancer cells escaping selection pressure imposed by the inhibitors. Shortly after ibrutinib was approved for CLL, mutations in the catalytic domain of BTK that confer resistance to ibrutinib were found. Moreover, activating mutations in PLCG2 were found that compensate for loss of BTK activity, as PLCγ2 is phosphorylated and activated by BTK (see figure).4
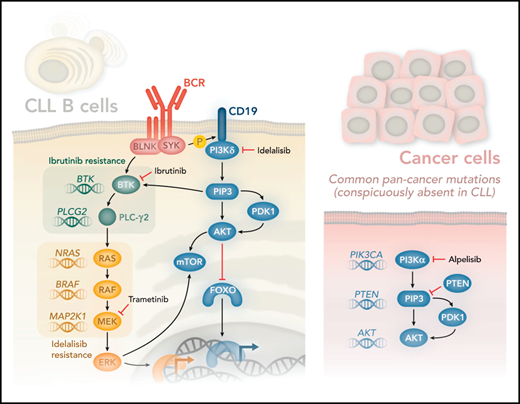
PI3Kδ is activated after SYK-mediated phosphorylation of CD19, which provides docking sites for PI3Kδ. PI3Kδ generates the lipid second-messenger molecule phosphatidylinositol(3,4,5)P3 (PIP3), which binds the PH domains of AKT and BTK. BTK is also regulated directly via SYK-mediated phosphorylation of BLNK. The activation of PLC-γ2 downstream of BTK is essential for generating the second-messenger molecules diacylglycerol and Ca2+ (not shown), which in turn control various transcription factors and contribute to the activation of RAS. RAS is at the apex of the MAPK signaling pathway, which is controlled by sequential activation of the kinases RAF, MEK, and ERK. Remarkably, Murali et al found activating mutations in genes encoding RAS, RAF, and MEK (mutated genes shown in italics in the figure). Strikingly, they found no mutations in genes typically associated with increased PI3K signaling in other cancer types (PIK3CA, PTEN, and AKT1-2, and -3). A search of 1308 CLL samples from The Cancer Genome Atlas returned no significant mutation in any of these genes (K.O., unpublished observations). Murali and colleagues therefore propose that PI3Kδ inhibitors such as idelalisib be combined with MAPK inhibitors, such as the MEK inhibitor trametinib, to circumvent adaptive resistance to therapy. Professional illustration by Somersault18:24.
PI3Kδ is activated after SYK-mediated phosphorylation of CD19, which provides docking sites for PI3Kδ. PI3Kδ generates the lipid second-messenger molecule phosphatidylinositol(3,4,5)P3 (PIP3), which binds the PH domains of AKT and BTK. BTK is also regulated directly via SYK-mediated phosphorylation of BLNK. The activation of PLC-γ2 downstream of BTK is essential for generating the second-messenger molecules diacylglycerol and Ca2+ (not shown), which in turn control various transcription factors and contribute to the activation of RAS. RAS is at the apex of the MAPK signaling pathway, which is controlled by sequential activation of the kinases RAF, MEK, and ERK. Remarkably, Murali et al found activating mutations in genes encoding RAS, RAF, and MEK (mutated genes shown in italics in the figure). Strikingly, they found no mutations in genes typically associated with increased PI3K signaling in other cancer types (PIK3CA, PTEN, and AKT1-2, and -3). A search of 1308 CLL samples from The Cancer Genome Atlas returned no significant mutation in any of these genes (K.O., unpublished observations). Murali and colleagues therefore propose that PI3Kδ inhibitors such as idelalisib be combined with MAPK inhibitors, such as the MEK inhibitor trametinib, to circumvent adaptive resistance to therapy. Professional illustration by Somersault18:24.
The PI3K pathway is among the most frequently perturbed pathways in cancer.5 B cells primarily express the PI3Kα and PI3Kδ isoforms. Although PI3Kδ is dominant, PI3Kα can compensate in part for the loss of PI3Kδ in B cells.6 Idelalisib binds the adenosine triphosphate pocket of PI3Kδ, but no mutations have been described that prevent such binding and hence could explain resistance. Theoretically, activating mutation in PIK3CA could have compensated, because activated PI3Kα is not inhibited by idelalisib. Moreover, PIK3CA is the most commonly mutated kinase in cancers, although, for unknown reasons, not in CLL. Similarly, PTEN, which antagonizes PI3K signaling, is among the most frequently mutated tumor-suppressor genes in cancer (next only to p53), but again, not in CLL. Congenital mutations in PIK3CD are associated with the primary immune deficiency syndrome activated PI3K-δ syndrome and sometimes also somatically in diffuse large B-cell lymphoma,7 but not in CLL (see figure). Hence, although CLL cells are highly dependent on signaling via the PI3K pathway, as evidenced by the marked clinical response to idelalisib, CLL is among the few cancer types that is rarely, if ever, caused by mutations in the PI3K pathway. How, then, do patients with CLL develop resistance to PI3K inhibitors?
To approach this question, Murali and colleagues sequenced CLL cells from patients who either did or did not respond to treatment with idelalisib. Among the nonresponders they confirmed a lack of mutations that could lead to reactivation of the PI3K pathway. Instead, they found several mutations that could activate the parallel MAPK pathway, as evidenced by mutations in 3 genes in this pathway: NRAS, BRAF, and MAP2K1 (see figure). In nontransformed B cells, PI3K signaling can contribute to the activation of the MAPK pathway, as indicated by phosphorylation of extracellular signal-regulated kinase (ERK), possibly by contributing to upstream BTK activation. However, in cells from patients with idelalisib-resistant disease, ERK phosphorylation was no longer sensitive to inhibition by idelalisib, but was still sensitive to inhibitors that target the MAPK pathway, including trametinib, a MEK inhibitor that is approved for the treatment of BRAF-mutant melanoma.8 By contrast, phosphorylation of AKT, which is an indicator of PI3K activity, was similarly inhibited by idelalisib in cells from patients with both resistant and susceptible disease, consistent with the lack of compensatory mutations in the PI3K pathway. Moreover, treating cells with idelalisib and MEK inhibitors led to enhanced inhibition of cell survival and proliferation. These results therefore suggest that the addition of an MAPK pathway inhibitor can overcome adaptive resistance to idelalisib.
Patients with CLL now have several novel therapies available to them. In addition to idelalisib and ibrutinib, venetoclax, which blocks BCL2 function, has been approved.9 Idelalisib has been plagued with adverse effects, including colitis and pneumonitis.10 There may therefore be little appetite for combining idelalisib with another inhibitor that can add to the list of adverse effects. However, second-generation inhibitors potentially with higher selectivity and/or fewer adverse effects are also being developed, and several have now been approved.11 Moreover, emerging evidence suggest that PI3Kδ inhibitors may be used intermittently and may reduce adverse effects while retaining therapeutic effects. Following this logic, sequential use of inhibitors may also be an attractive option. This approach would require new clinical trials, as MAPK inhibitors have not yet been approved for CLL.
Why are mutations activating this pathway not found in CLL when they are so common in other cancers? Why is drug resistance not associated with such mutations? Generating hypotheses based on the absence of evidence is a challenge, but these 2 studies are starting to suggest that, although normal PI3K signaling is essential for CLL, hyperactivated PI3K signaling may be selected against. Moreover, if CLL cells can compensate with PI3K inhibition by increasing MAPK signaling, does it mean that PI3K-δ–dependent MAPK activation is essential for CLL? The other possibility is that the PI3K and MAPK pathways share a common target. In this context, it is worth noting that both the PI3K and MAPK pathways can converge after activation of mTOR and its downstream substrate, S6K.
Both BTK and PI3Kδ inhibition act in part by purging CLL cells from their protective interactions with the stroma in the lymph nodes. This action leads to the characteristic lymphocytosis observed shortly after treatment and renders the CLL cells more susceptible to apoptosis, which can be accelerated by coadministered therapeutics, such as rituximab or chemotherapy.2 At present, it is not known whether the MAPK pathway can also trigger lymphocytosis, but this possibility should be monitored closely in future trials.
The results in Murali et al highlight a key role for MAPK pathway activation as a resistance mechanism, suggesting that MAPK pathway inhibitors may be considered for patients who no longer respond to PI3K-δ inhibitors.
Conflict-of-interest disclosure: K.O. has consulted for and received speaker fees from Gilead Sciences Inc.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal