

Key Points
NETs contribute to cerebrovascular complications of tPA via cGAS-STING activation and type 1 interferon response in ischemic brain.
Targeting NETs or cGAS improves thrombolytic therapy by blocking tPA-associated BBB breakdown and hemorrhage.

Abstract
Intracerebral hemorrhage associated with thrombolytic therapy with tissue plasminogen activator (tPA) in acute ischemic stroke continues to present a major clinical problem. Here, we report that infusion of tPA resulted in a significant increase in markers of neutrophil extracellular traps (NETs) in the ischemic cortex and plasma of mice subjected to photothrombotic middle cerebral artery occlusion. Peptidylarginine deiminase 4 (PAD4), a critical enzyme for NET formation, is also significantly upregulated in the ischemic brains of tPA-treated mice. Blood–brain barrier (BBB) disruption after ischemic challenge in an in vitro model of BBB was exacerbated after exposure to NETs. Importantly, disruption of NETs by DNase I or inhibition of NET production by PAD4 deficiency restored tPA-induced loss of BBB integrity and consequently decreased tPA-associated brain hemorrhage after ischemic stroke. Furthermore, either DNase I or PAD4 deficiency reversed tPA-mediated upregulation of the DNA sensor cyclic GMP-AMP (cGAMP) synthase (cGAS). Administration of cGAMP after stroke abolished DNase I–mediated downregulation of the STING pathway and type 1 interferon production and blocked the antihemorrhagic effect of DNase I in tPA-treated mice. We also show that tPA-associated brain hemorrhage after ischemic stroke was significantly reduced in cGas−/− mice. Collectively, these findings demonstrate that NETs significantly contribute to tPA-induced BBB breakdown in the ischemic brain and suggest that targeting NETs or cGAS may ameliorate thrombolytic therapy for ischemic stroke by reducing tPA-associated hemorrhage.
Introduction
Thrombolysis with tissue plasminogen activator (tPA) is the only approved pharmacological therapy for acute ischemic stroke1,2 because of its thrombolytic effect.3 However, the efficacy of tPA therapy is counteracted by an increased risk of intracerebral hemorrhage.4,5 Although there is a clear need to identify an adjuvant agent for increasing the safety of tPA thrombolysis for ischemic stroke, the contributing factors leading to hemorrhage are still not fully understood.
tPA can activate the brain endothelium, which results in the degradation of vascular integrity and acute blood–brain barrier (BBB) breakdown.6,7 The infiltration of neutrophils from the microvasculature into the brain parenchyma represents a key event during BBB leakage after ischemic stroke.8 Interestingly, tPA was shown to promote the recruitment of neutrophils to ischemic tissue.9,10 Once activated, neutrophils can release neutrophil extracellular traps (NETs).11,12 NETs are extracellular DNA fibers consisting of histones and granular proteins.13 They have been identified as major triggers of thrombosis and inflammation.11,14 Recently, NETs were detected in the thrombi of patients with ischemic stroke.15 Disruption of NETs by DNase I reduced ischemic brain injury and improved tPA-induced thrombolysis. Our recent work also indicates that targeting NETs improves vascular remodeling during stroke recovery.16
In this study, we sought to investigate the effects of tPA on the generation of NETs in a mouse model of thrombotic middle cerebral artery occlusion (MCAO) and determine whether targeting NETs via DNase I or peptidylarginine deiminase 4 (PAD4) deficiency could increase the safety of tPA thrombolysis in ischemic stroke. We show that the release of NETs impairs cerebrovascular integrity and exacerbates tPA-induced intracerebral hemorrhage. We also uncovered a crucial pathway for NETs in regulating BBB permeability and cerebral hemorrhage associated with tPA treatment involving the cyclic GMP-AMP (cGAMP) synthase (cGAS)–STING axis and its downstream signaling molecules.
Methods
Photothrombotic stroke model
Pad4−/− and cGas−/− mice on a C57BL/6 background were purchased from The Jackson Laboratory. Male wild-type (WT; C57BL/6) mice (age 8-10 weeks) were purchased from Shanghai Laboratory Animal Center (SLAC) Laboratory Animal Co, Ltd (Shanghai, China). Animal protocols were approved by the Animal Care and Use Committee of Shanghai Medical College, Fudan University. Photothrombotic MCAO was induced as described.17,18 Mice were anesthetized by 1% to 1.5% isoflurane in 30% oxygen and 70% nitrous oxide. Body temperature was maintained at 37°C ± 0.5°C using a temperature control unit. The skin between the right eye and right ear was incised, and the temporal muscle was retracted. In the MCA region, a window 1.5 mm in diameter was opened using a high-speed micro drill (Stoelting; CellPoint Scientific, Gaithersburg, MD), and the dura mater was intact. Irradiation with green laser (3.5 mW; wavelength, 540 nm; AniLab Software and Instruments, Ningbo, China) was directed using an optic fiber 200 μm in diameter. The photosensitive dye Rose Bengal (50 mg/kg; Sigma-Aldrich, St Louis, MO) was injected through the tail vein, and the MCA was illuminated for 5 minutes. After closing of the surgical wounds, mice were returned to their cages. d-Glucose (6 mL/kg at 50% weight/volume) was injected intraperitoneally 15 minutes before MCAO. At 2 hours after MCAO, human recombinant tPA (10 mg/kg; alteplase; Boehringer Ingelheim, Mannheim, Germany) or saline was administered as an IV bolus injection of 1 mg/kg followed by a 9 mg/kg continuous infusion for 30 minutes with an infusion pump (World Precision Instruments, Sarasota, FL).19 Human recombinant DNase I (enz-319-10000IU; ProSpec, Rehovot, Israel) or vehicle (8.77 mg/mL of sodium chloride and 0.15 mg/mL of calcium chloride) was administered by IV injection of 10 μg and intraperitoneal injection of 50 μg 15 minutes before tPA infusion and was then administered again 11 hours after the first injections.12 Ten minutes before tPA infusion, 2′3′-cGAMP (tlrl-nacga23-1; InvivoGen, San Diego, CA) or vehicle was delivered at an IV dose of 1 mg/kg; this was repeated 11 hours after the first injection.20 Receptor-associated protein (RAP; 1 mg/kg; Sigma-Aldrich),21 tranexamic acid (300 mg/kg; Sigma-Aldrich),22 or saline was administered by IV injection 10 minutes before tPA infusion.
Neutrophil isolation and in vitro NET assay
Neutrophils were isolated from whole blood or bone marrow using Percoll (GE Healthcare) gradient separation as described.16,23 Peripheral blood neutrophils were seeded at a density of 5 × 105 cells per mL in 48-well glass-bottomed plates and treated with tPA or 10 μg/mL Klebsiella pneumoniae lipopolysaccharide (LPS; Sigma-Aldrich) for 2.5 hours at 37°C.16 Cl-amidine (100 μM; Millipore, Burlington, MA),24 RAP (500 nM),25 or vehicle was added 15 minutes before addition of tPA. Cells were fixed in 2% paraformaldehyde, blocked with 3% bovine serum albumin, and incubated with rabbit anti-citrullinated histone H3 (H3Cit; 1:1000; ab5103; Abcam, Cambridge, MA), followed by incubation with Alexa Fluor 488 donkey anti-rabbit secondary antibody. Hoechst 33342 (1:10 000; H3570; Invitrogen, Waltham, MA) was used for DNA detection.
NET preparations
Neutrophils isolated from bone marrow were suspended in RPMI 1640 (Gibco, Waltham, MA) and seeded in 6-well plates at a density of 5 × 105 cells per mL. Neutrophils were stimulated with 100 nM of phorbol 12-myristate 13-acetate (Sigma-Aldrich) for 4 hours at 37°C in a humidified 5% carbon dioxide (CO2) incubator.26 After removing the medium, wells were washed twice with phosphate-buffered saline. NETs were collected in 15-mL tubes by vigorous agitation. After centrifugation at 300g for 5 minutes, DNA concentration of supernatants containing NETs was quantified using the Picogreen dsDNA Kit (Invitrogen).
In vitro model of the BBB
Human brain microvascular endothelial cells (HBMECs; Sciencell, Carlsbad, CA) between passages 3 and 8 were grown in EGM-2 MV medium (Lonza, Walkersville, MD). Transwell membrane inserts (0.4-μm pores, 11-mm diameter; Corning Life Science, Lowell, MA) were coated with collagen IV (50 μg/mL) and fibronectin (30 μg/mL).27 HBMECs (density, 2 × 105 cells) were seeded onto the upper side of the insert. Cultures were maintained at 37°C in a humidified 95% air and 5% CO2 incubator for 4 days. Ischemia was achieved using an oxygen-glucose deprivation (OGD) condition.28 After replacing cell culture medium with glucose-free medium, cultures were exposed to 2 hours of hypoxia (95% nitrogen and 5% CO2) at 37°C using a hypoxia chamber (Billups-Rothenberg, Del Mar, CA). After OGD, cells were incubated in medium containing glucose, and cultures were placed into 95% air and 5% CO2. NETs (1.5 μg/mL) were added immediately after OGD.29 For cell permeability assay, 40 KDa of fluorescein isothiocyanate (FITC)–dextran (Sigma-Aldrich; 0.5 mL of 2 mg/mL of fresh medium) was added to the upper side of the insert. Fluorescence intensity of FITC-dextran in the lower chamber was measured every hour for 6 hours using a fluorescent microplate reader (BioTek Instruments, Inc, Winooski, VT). The concentration of FITC-dextran in samples was calculated from a standard curve generated using known concentrations of tracer.30
In vivo multiphoton microscopy analysis of BBB permeability
Cranial windows were prepared as we previously described.31,32 Briefly, mice were anesthetized with 1% to 1.5% isoflurane in 30% oxygen and 70% nitrous oxide and kept on a heating plate (37°C ± 0.5°C). After fixation in a custom-made head holder, a window 2 mm in diameter was made in the parietal bone (centered 2.5 mm lateral and 1 mm posterior to the bregma) using a high-speed micro drill (Stoelting). A sterile 5 × 5 mm cover glass (World Precision Instruments) was placed above the window and fixed with dental cement. For in vivo imaging, an Olympus FluoView FVMPE-RS upright multiphoton laser-scanning system mounted on an Olympus XL Plan N 25×/1.05 WMP ∞/0-0.23/FN/18 dipping objective was used. Two-photon excitation was performed with Mai Tai eHPDS-OL (Santa Clara, CA) and Spectra Physics InSight DS-OL lasers (Santa Clara, CA). Emitted fluorescence was detected through a 495- to 540-nm band pass filter. To analyze cortical cerebrovascular permeability, FITC-dextran (molecular weight, 40 KDa; Sigma-Aldrich; 0.1 mL of 10 mg/mL) was injected IV, and time lapse imaging of FITC-dextran was acquired every 3 minutes for 30 minutes. The fluorescence of randomly chosen 20 × 20 μm2 regions of interest within the vessel and corresponding areas within the perivascular brain parenchyma was recorded as described.16
Quantification of cerebral hemorrhage
At 24 hours after MCAO, mice were perfused transcardially with phosphate-buffered saline. Drabkin reagent (500 μL; Sigma-Aldrich) was added to the ischemic hemispheric brain tissue of each mouse, followed by homogenization and centrifugation at 13 000 rpm for 30 minutes. Optical density of supernatants was measured at 540 nm. Hemorrhage volume was expressed in equivalent units by comparison with a reference curve generated using fresh homologous blood as described.19
Measurements of DNA in plasma, IL-6, and IFN-β levels in brain tissue
Plasma collected from whole blood was separated by centrifugation. Plasma DNA was quantified using the Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen). Cortical tissues were lysed in RIPA lysis buffer (Millipore) including protease inhibitor cocktails (Roche Diagnostics, GmbH, Mannheim, Germany). Interleukin-6 (IL-6) and interferon-β (IFN-β) levels were measured by enzyme-linked immunosorbent assay (R&D Systems; PBL Assay Science) according to the manufacturer’s guidelines.
Statistical analysis
Data are presented as means ± standard deviations and were analyzed using GraphPad Prism 7 software. Multiple comparisons were performed using 1-way analysis of variance followed by Tukey’s multiple comparison test. When comparing 2 groups, an unpaired Student t test was used. A value of P < .05 was considered statistically significant.
Results
tPA activates neutrophils to overproduce NETs after ischemic stroke
Infusion of tPA 2 hours after MCAO resulted in a substantial increase in neutrophil recruitment to the ischemic cortex compared with vehicle-treated mice (Figure 1A-B). Counting the neutrophils in immunostained brain sections also indicated that tPA treatment increased neutrophil counts in the peri-infarct cortex by ∼96% when compared with the vehicle group (Figure 1C; supplemental Figure 1A). Thereafter, consistent with these data, stroke-induced upregulation of the neutrophil enzyme MPO was significantly increased by tPA treatment (Figure 1D).
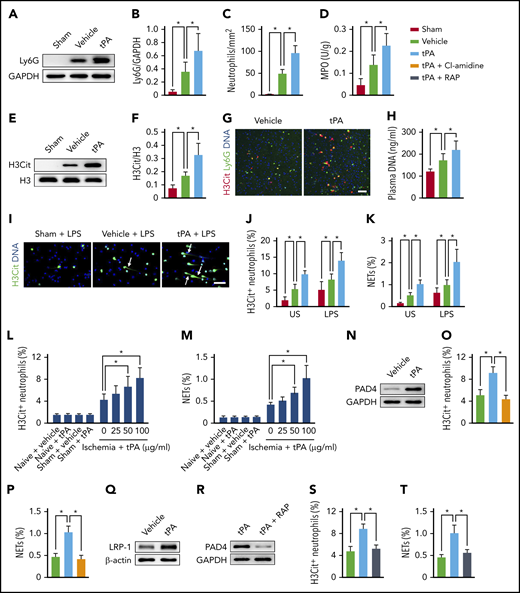
tPA activates neutrophils to release NETs after ischemic stroke. (A-B) Representative immunoblots and quantitative determinations of the amount of neutrophils in the ischemic cortex at 24 hours after stroke in mice treated with vehicle or tPA compared with mice undergoing sham surgery (n = 5). (C) Quantification of the numbers of infiltrating neutrophils in the ischemic cortex at 24 hours after stroke (n = 6). (D) Quantification of MPO activity in the ischemic brain (n = 6). (E-F) Representative immunoblots and quantitative determinations of H3Cit levels in the ischemic cortex (n = 5). (G) Representative confocal images of H3Cit+ neutrophils in the peri-infarct cortex. Scale bar, 50 μm. (H) Quantification of plasma DNA (n = 6). (I) Representative images of isolated peripheral blood neutrophils from mice undergoing sham surgery and ischemic mice treated with vehicle or tPA. Neutrophils were incubated with LPS for 2.5 hours. DNA was stained with Hoechst 33342 (blue), and neutrophils were stained with H3Cit (green). Arrows indicate NETs. Scale bar, 50 μm. (J-K) Quantification of the percentage of H3Cit+ neutrophils and NETs in unstimulated (US) and LPS-stimulated peripheral blood neutrophils (n = 8). (L-M) Quantification of the percentage of H3Cit+ neutrophils and NETs in peripheral blood neutrophils isolated from naïve mice, mice undergoing sham surgery, or ischemic mice (n = 5-8). Neutrophils from naive mice or mice undergoing sham surgery were treated with vehicle or 100 μg/mL of tPA. Neutrophils from ischemic mice were treated with vehicle or 25, 50, or 100 μg/mL of tPA. (N) Representative immunoblots of PAD4 expression in neutrophils isolated from ischemic mice. Neutrophils were treated with vehicle or 100 μg/mL of tPA. (O-P) Quantification of the percentage of H3Cit+ neutrophils and NETs in neutrophils isolated from ischemic mice (n = 8). Neutrophils were treated with vehicle, 100 μg/mL of tPA, or tPA in combination with the PAD inhibitor Cl-amidine. (Q) Representative immunoblots of lipoprotein receptor–related protein 1 (LRP-1) expression in neutrophils isolated from ischemic mice. Neutrophils were treated with vehicle or 100 μg/mL of tPA. (R) Representative immunoblots of PAD4 expression in neutrophils isolated from ischemic mice. Neutrophils were treated with 100 μg/mL of tPA or tPA in combination with the LRP antagonist RAP. (S-T) Quantification of the percentage of H3Cit+ neutrophils and NETs in neutrophils isolated from ischemic mice (n = 8). Neutrophils were treated with vehicle, 100 μg/mL of tPA, or tPA in combination with the LRP antagonist RAP. Values are means ± standard deviation. *P < .05. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
tPA activates neutrophils to release NETs after ischemic stroke. (A-B) Representative immunoblots and quantitative determinations of the amount of neutrophils in the ischemic cortex at 24 hours after stroke in mice treated with vehicle or tPA compared with mice undergoing sham surgery (n = 5). (C) Quantification of the numbers of infiltrating neutrophils in the ischemic cortex at 24 hours after stroke (n = 6). (D) Quantification of MPO activity in the ischemic brain (n = 6). (E-F) Representative immunoblots and quantitative determinations of H3Cit levels in the ischemic cortex (n = 5). (G) Representative confocal images of H3Cit+ neutrophils in the peri-infarct cortex. Scale bar, 50 μm. (H) Quantification of plasma DNA (n = 6). (I) Representative images of isolated peripheral blood neutrophils from mice undergoing sham surgery and ischemic mice treated with vehicle or tPA. Neutrophils were incubated with LPS for 2.5 hours. DNA was stained with Hoechst 33342 (blue), and neutrophils were stained with H3Cit (green). Arrows indicate NETs. Scale bar, 50 μm. (J-K) Quantification of the percentage of H3Cit+ neutrophils and NETs in unstimulated (US) and LPS-stimulated peripheral blood neutrophils (n = 8). (L-M) Quantification of the percentage of H3Cit+ neutrophils and NETs in peripheral blood neutrophils isolated from naïve mice, mice undergoing sham surgery, or ischemic mice (n = 5-8). Neutrophils from naive mice or mice undergoing sham surgery were treated with vehicle or 100 μg/mL of tPA. Neutrophils from ischemic mice were treated with vehicle or 25, 50, or 100 μg/mL of tPA. (N) Representative immunoblots of PAD4 expression in neutrophils isolated from ischemic mice. Neutrophils were treated with vehicle or 100 μg/mL of tPA. (O-P) Quantification of the percentage of H3Cit+ neutrophils and NETs in neutrophils isolated from ischemic mice (n = 8). Neutrophils were treated with vehicle, 100 μg/mL of tPA, or tPA in combination with the PAD inhibitor Cl-amidine. (Q) Representative immunoblots of lipoprotein receptor–related protein 1 (LRP-1) expression in neutrophils isolated from ischemic mice. Neutrophils were treated with vehicle or 100 μg/mL of tPA. (R) Representative immunoblots of PAD4 expression in neutrophils isolated from ischemic mice. Neutrophils were treated with 100 μg/mL of tPA or tPA in combination with the LRP antagonist RAP. (S-T) Quantification of the percentage of H3Cit+ neutrophils and NETs in neutrophils isolated from ischemic mice (n = 8). Neutrophils were treated with vehicle, 100 μg/mL of tPA, or tPA in combination with the LRP antagonist RAP. Values are means ± standard deviation. *P < .05. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
We then asked whether tPA could contribute to neutrophil priming. Western blot analysis of the ischemic cortex indicated a marked increase in the levels of H3Cit in mice subjected to stroke (Figure 1E-F) as reported.8,16 Infusion of tPA in mice subjected to MCAO resulted in an even greater increase in the levels of H3Cit compared with mice treated with MCAO and vehicle. We then found a substantial 2.4-fold increase in H3Cit+ neutrophils in mice subjected to MCAO and tPA treatment compared with mice treated with MCAO and vehicle (Figure 1G; supplemental Figure 1B). In contrast, H3Cit+ neutrophils were almost undetectable in mice undergoing sham surgery. In line with this, elevated levels of circulating DNA were found in the plasma from mice treated with tPA (Figure 1H).
We then isolated neutrophils from the peripheral blood of mice undergoing sham surgery and ischemic mice treated with vehicle or tPA and incubated them with or without LPS stimulation. We found that either unstimulated or LPS-stimulated neutrophils from ischemic mice treated with tPA showed significant increases in H3Cit+ neutrophils and NET formation compared with mice undergoing sham surgery and ischemic mice treated with vehicle (Figure 1I-K). We also isolated neutrophils from the peripheral blood of naive mice, mice undergoing sham surgery, and ischemic mice and treated them with tPA or vehicle. Neutrophils from ischemic mice treated with tPA showed a dose-dependent increase in H3Cit+ neutrophils and NET formation compared with vehicle-treated neutrophils (Figure 1L-M). However, tPA did not induce more neutrophils from naive mice or those undergoing sham surgery to become H3Cit+ neutrophils or form NETs compared with vehicle-treated neutrophils, suggesting a possible priming role of ischemia. Together, these data indicate that tPA is capable of stimulating neutrophils from ischemic mice to produce NETs.
We next investigated the mechanism underlying tPA-mediated formation of NETs. Western blot analysis revealed a significant increase in PAD4 expression in tPA-treated neutrophils from ischemic mice compared with vehicle-treated neutrophils (Figure 1N; supplemental Figure 2A). Treatment with the PAD inhibitor Cl-amidine resulted in a significant decrease in H3Cit+ neutrophils (Figure 1O; supplemental Figure 2B) and NET formation (Figure 1P) in tPA-treated neutrophils. We then observed that tPA-treated neutrophils from ischemic mice exhibited an increase in the expression of the tPA receptor low-density LRP-1 (Figure 1Q; supplemental Figure 2C), which is consistent with findings from a recent in vitro study.25 Furthermore, the tPA-induced PAD4 expression (Figure 1R; supplemental Figure 2D), H3Cit+ neutrophils (Figure 1S; supplemental Figure 2E), and NET production (Figure 1T) were decreased when neutrophils were incubated with a combination of tPA and the LRP antagonist RAP.
We next asked whether RAP could reduce tPA-mediated neutrophil recruitment and NET formation after stroke in mice. Western blot analysis revealed that treatment with tPA significantly increased LRP-1 expression in the ischemic cortex compared with vehicle injection (Figure 2A-B). Immunostaining also confirmed increased LRP-1 expression in tPA-treated mice (Figure 2C). We then observed that inhibition of LRP-1 by RAP significantly reduced MPO activity and H3Cit levels in tPA-treated mice (Figure 2D-E; supplemental Figure 3A). Together, these data indicate that tPA contributes to neutrophil recruitment and directly induces NET formation under ischemic conditions and suggest that these effects may occur through LRP-1–mediated upregulation of PAD4.
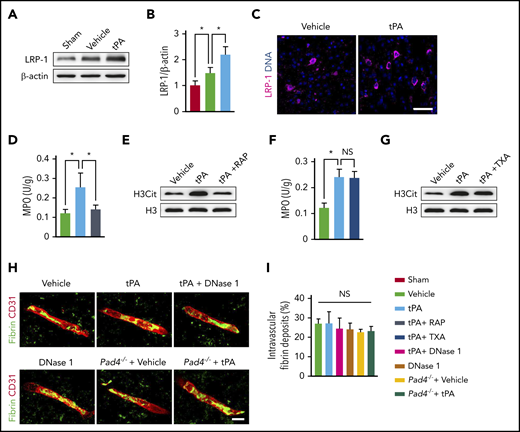
LRP-1 mediates the effects of tPA on neutrophil recruitment and NET formation after ischemic stroke. (A-B) Representative immunoblots and quantitative determinations of LRP-1 expression in the ischemic cortex at 24 hours after stroke in mice treated with vehicle or tPA compared with mice undergoing sham surgery (n = 5). (C) Representative confocal images of LRP-1 immunostaining in the ischemic cortex. DNA was stained with 4′,6-diamidino-2-phenylindole (blue). Independent experiments are repeated ≥3 times. Scale bar, 40 μm. (D) Quantification of MPO activity in the ischemic brain (n = 6). (E) Representative immunoblots of H3Cit in the ischemic cortex. (F) Quantification of MPO activity in the ischemic brain (n = 6). (G) Representative immunoblots of H3Cit in the ischemic cortex. (H) Representative confocal images of fibrin intravascular deposits (green) and CD31+ microvessels (red) in the infarct areas at 24 hours. Scale bar, 10 μm. (I) Quantification of fibrin intravascular deposits for each group (n = 6). Values are means ± standard deviation. *P < .05. NS, not significant; TXA, tranexamic acid.
LRP-1 mediates the effects of tPA on neutrophil recruitment and NET formation after ischemic stroke. (A-B) Representative immunoblots and quantitative determinations of LRP-1 expression in the ischemic cortex at 24 hours after stroke in mice treated with vehicle or tPA compared with mice undergoing sham surgery (n = 5). (C) Representative confocal images of LRP-1 immunostaining in the ischemic cortex. DNA was stained with 4′,6-diamidino-2-phenylindole (blue). Independent experiments are repeated ≥3 times. Scale bar, 40 μm. (D) Quantification of MPO activity in the ischemic brain (n = 6). (E) Representative immunoblots of H3Cit in the ischemic cortex. (F) Quantification of MPO activity in the ischemic brain (n = 6). (G) Representative immunoblots of H3Cit in the ischemic cortex. (H) Representative confocal images of fibrin intravascular deposits (green) and CD31+ microvessels (red) in the infarct areas at 24 hours. Scale bar, 10 μm. (I) Quantification of fibrin intravascular deposits for each group (n = 6). Values are means ± standard deviation. *P < .05. NS, not significant; TXA, tranexamic acid.
We further studied whether plasmin is required for the tPA-mediated effects on neutrophil activity after stroke. We found that treatment with tranexamic acid in combination with tPA did not change MPO activity or H3Cit levels compared with mice treated with tPA alone (Figure 2F-G; supplemental Figure 3B), suggesting that the tPA-mediated effects on neutrophil activity were independent of plasmin. In addition, we observed that neither DNase I treatment nor PAD4 deficiency affected intravascular fibrin deposits in the infarct areas in mice subjected to MCAO or MCAO plus tPA (Figure 2H-I).
Clearance of NETs by DNase I or inhibition of NET generation by PAD4 deficiency protects BBB integrity and blocks tPA-induced hemorrhage
We first studied the role of NETs in stroke-induced BBB disruption by using an in vitro BBB model composed of a monolayer of HBMECs. Brain endothelial monolayers were subjected to 2 hours of OGD followed by NET treatment. Addition of NETs significantly exacerbated the OGD-induced endothelial hyperpermeability to dextran (Figure 3A).
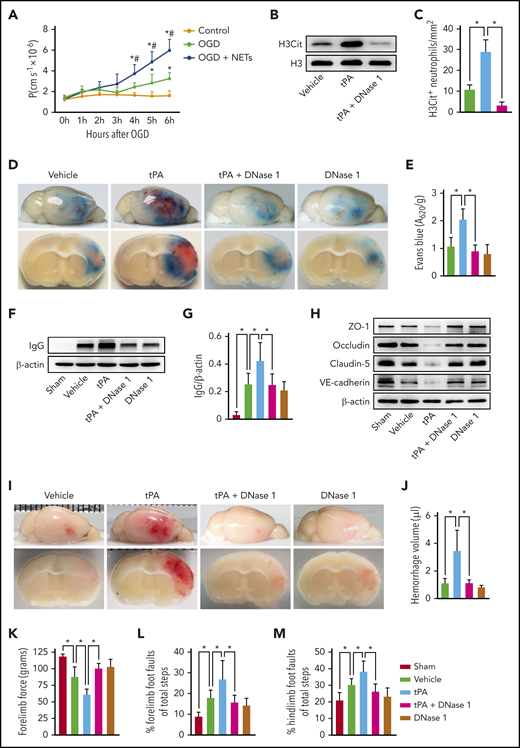
DNase I treatment reduces tPA-mediated BBB breakdown and cerebral hemorrhage after ischemic stroke. (A) Permeability coefficient (P) of human brain endothelial monolayers to 40 KDa of FITC-dextran at 0 to 6 hours after normoxia or OGD with or without NETs (1.5 μg/mL). (B) Representative immunoblots of H3Cit levels in the ischemic cortex at 24 hours after stroke in mice treated with vehicle, tPA, or tPA in combination with DNase I. (C) Quantification of the numbers of H3Cit+ neutrophils in the ischemic cortex (n = 6). (D) Representative images of the dorsal surface (upper panel) and a coronal section (bottom panel) show Evans blue extravasation 24 hours after stroke in mice treated with vehicle, tPA, tPA in combination with DNase I, or DNase I alone. (E) Quantification of Evans blue fluorescence intensity for each group (n = 8). (F-G) Representative immunoblots and quantification of immunoglobulin G (IgG) levels in capillary-depleted brain tissue at 24 hours after stroke (n = 5). (H) Representative immunoblots of the tight junction proteins zonula occludens-1 (ZO-1), claudin-5, and occludin and the adherens junction protein vascular endothelial–cadherin (VE-cadherin) in isolated brain microvessels. (I) Representative images of the dorsal surface (upper panel) and a coronal section (bottom panel) show cerebral hemorrhage 24 hours after stroke in mice treated with vehicle, tPA, tPA in combination with DNase I, or DNase I alone. (J) Quantification of cerebral hemorrhage by spectrophotometric hemoglobin assay (n = 8). (K-M) DNase I treatment improved neurological functions in forelimb force test and beam walking test (n = 10). Values are means ± standard deviation. *P < .05 vs control group (A), #P < .05 vs OGD group (A), *P < .05 (C,E,G,J-M).
DNase I treatment reduces tPA-mediated BBB breakdown and cerebral hemorrhage after ischemic stroke. (A) Permeability coefficient (P) of human brain endothelial monolayers to 40 KDa of FITC-dextran at 0 to 6 hours after normoxia or OGD with or without NETs (1.5 μg/mL). (B) Representative immunoblots of H3Cit levels in the ischemic cortex at 24 hours after stroke in mice treated with vehicle, tPA, or tPA in combination with DNase I. (C) Quantification of the numbers of H3Cit+ neutrophils in the ischemic cortex (n = 6). (D) Representative images of the dorsal surface (upper panel) and a coronal section (bottom panel) show Evans blue extravasation 24 hours after stroke in mice treated with vehicle, tPA, tPA in combination with DNase I, or DNase I alone. (E) Quantification of Evans blue fluorescence intensity for each group (n = 8). (F-G) Representative immunoblots and quantification of immunoglobulin G (IgG) levels in capillary-depleted brain tissue at 24 hours after stroke (n = 5). (H) Representative immunoblots of the tight junction proteins zonula occludens-1 (ZO-1), claudin-5, and occludin and the adherens junction protein vascular endothelial–cadherin (VE-cadherin) in isolated brain microvessels. (I) Representative images of the dorsal surface (upper panel) and a coronal section (bottom panel) show cerebral hemorrhage 24 hours after stroke in mice treated with vehicle, tPA, tPA in combination with DNase I, or DNase I alone. (J) Quantification of cerebral hemorrhage by spectrophotometric hemoglobin assay (n = 8). (K-M) DNase I treatment improved neurological functions in forelimb force test and beam walking test (n = 10). Values are means ± standard deviation. *P < .05 vs control group (A), #P < .05 vs OGD group (A), *P < .05 (C,E,G,J-M).
We next subjected mice to photothrombotic MCAO to determine the role of NETs in BBB damage in the setting of cerebral ischemia. To degrade NETs, we treated mice with DNase I, which has been shown to dissolve NETs. Removal of NETs (Figure 3B-C; supplemental Figure 4A) with DNase I significantly reduced the extravasation of Evans blue dye in tPA-treated mice (Figure 3D-E). Immunoblotting of microvessel-depleted brain cortex showed that DNase I also reduced the leakage of endogenous serum IgG (Figure 3F-G). Consistent with these findings, several junctional proteins, including tight junction proteins zonula occludens-1, occludin, claudin-5, and adherens junction protein vascular endothelial–cadherin, which are required for BBB integrity,33,34 were normalized in brain microvessels of tPA-treated mice by DNase I (Figure 3H; supplemental Figure 5A-D).
To test whether DNase I could influence the incidence of tPA-associated cerebral hemorrhage, we analyzed hemorrhage volume at 24 hours after stroke. We found that infusion of tPA produced a more than threefold increase in cerebral hemorrhage compared with vehicle-treated mice (Figure 3I-J). DNase I in combination with tPA blocked tPA-induced hemorrhage. tPA alone worsened the neurological deficits as assayed by the forelimb force test and beam walking test (Figure 3K-M). In contrast, DNase I improved the neurological deficits in tPA-treated mice.
PAD4 is an essential enzyme for NET formation that catalyzes the citrullination of histone H3.35,36 Western blotting of brain tissue indicated a marked upregulation of PAD4 expression in mice subjected to MCAO as compared with mice undergoing sham surgery (Figure 4A-B). We then observed a sustained 2.2-fold increase in PAD4 expression in mice subjected to MCAO plus tPA compared with mice treated with MCAO and vehicle. Therefore, we tested the effect of PAD4 deficiency on tPA-induced BBB disruption and cerebral hemorrhage in mice with stroke. We found that the levels of H3Cit in the ischemic brain were lowered by 88% in tPA-treated Pad4−/− mice (supplemental Figure 6A-B), whereas neutrophil infiltration was not affected (supplemental Figure 6C-D). Mice deficient in PAD4 treated with tPA exhibited substantial reductions in Evans blue dye extravasation (Figure 4C-D), perivascular IgG deposits (Figure 4E-F), and loss of zonula occludens-1, occludin, claudin-5, and vascular endothelial–cadherin (Figure 4G-K) after ischemic stroke compared with tPA-treated WT mice. Furthermore, tPA-treated Pad4−/− mice showed a marked 68% reduction in cerebral hemorrhage (Figure 4L-M) and improvement in neurological deficits (Figure 4N-P) compared with tPA-treated WT mice.
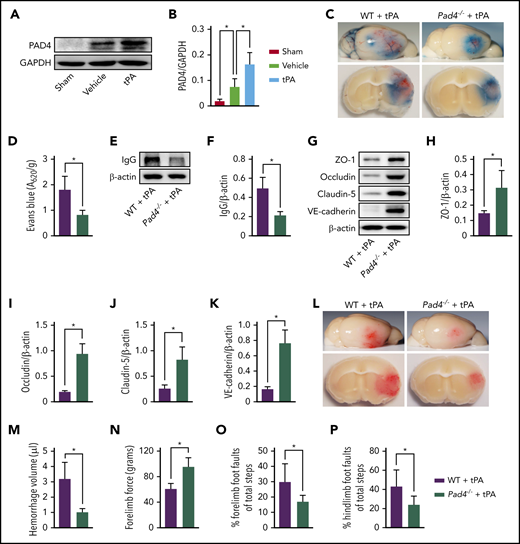
NETs and PAD4 contribute to tPA-mediated BBB disruption and cerebral hemorrhage after ischemic stroke. (A-B) Representative immunoblots and quantification of PAD4 levels in the ischemic cortex 24 hours after stroke in mice treated with vehicle or tPA compared with mice undergoing sham surgery (n = 5). (C) Representative images of the dorsal surface (upper panel) and a coronal section (bottom panel) show Evans blue extravasation 24 hours after stroke in WT and Pad4−/− mice treated with tPA. (D) Quantification of Evans blue fluorescence intensity for each group (n = 8). (E-F) Representative immunoblots and quantification of IgG levels in capillary-depleted brain tissue at 24 hours (n = 5). (G-K) Representative immunoblots and quantification of zonula occludens-1 (ZO-1), occludin, claudin-5, and vascular endothelial–cadherin (VE-cadherin) in isolated brain microvessels (n = 5). (L) Representative images of the dorsal surface (upper panel) and a coronal section (bottom panel) show cerebral hemorrhage 24 hours after stroke in WT and Pad4−/− mice treated with tPA. (M) Quantification of cerebral hemorrhage by spectrophotometric hemoglobin assay (n = 8). (N-P) Effects of PAD deficiency on forelimb force test and beam walking test 24 hours after stroke (n = 10). Values are means ± standard deviation. *P < .05. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
NETs and PAD4 contribute to tPA-mediated BBB disruption and cerebral hemorrhage after ischemic stroke. (A-B) Representative immunoblots and quantification of PAD4 levels in the ischemic cortex 24 hours after stroke in mice treated with vehicle or tPA compared with mice undergoing sham surgery (n = 5). (C) Representative images of the dorsal surface (upper panel) and a coronal section (bottom panel) show Evans blue extravasation 24 hours after stroke in WT and Pad4−/− mice treated with tPA. (D) Quantification of Evans blue fluorescence intensity for each group (n = 8). (E-F) Representative immunoblots and quantification of IgG levels in capillary-depleted brain tissue at 24 hours (n = 5). (G-K) Representative immunoblots and quantification of zonula occludens-1 (ZO-1), occludin, claudin-5, and vascular endothelial–cadherin (VE-cadherin) in isolated brain microvessels (n = 5). (L) Representative images of the dorsal surface (upper panel) and a coronal section (bottom panel) show cerebral hemorrhage 24 hours after stroke in WT and Pad4−/− mice treated with tPA. (M) Quantification of cerebral hemorrhage by spectrophotometric hemoglobin assay (n = 8). (N-P) Effects of PAD deficiency on forelimb force test and beam walking test 24 hours after stroke (n = 10). Values are means ± standard deviation. *P < .05. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
NETs are essential for tPA-induced activation of the cGAS-STING pathway
A major sensor for cytosolic DNA is cGAS, which leads to activation of STING and induction of type 1 IFN expression and IFN-stimulated proinflammatory cytokines.37,38 We observed a sustained increase in the expression of cGAS in the ischemic cortex in mice subjected to MCAO compared with mice undergoing sham surgery (Figure 5A-B). tPA further increased cGAS expression in this model, whereas this effect of tPA was abolished by DNase I. We then found that tPA amplified stroke-induced activation of STING, TANK-binding kinase 1 (TBK1), IFN regulatory factor 3 (IRF-3; Figure 5C-F), and IFN-β and IL-6 induction (Figure 5G-H). DNase I and deficiency of PAD4 substantially reduced the activation of the cGAS-STING pathway and the production of IFN-β and IL-6 in mice treated with tPA and ischemia.
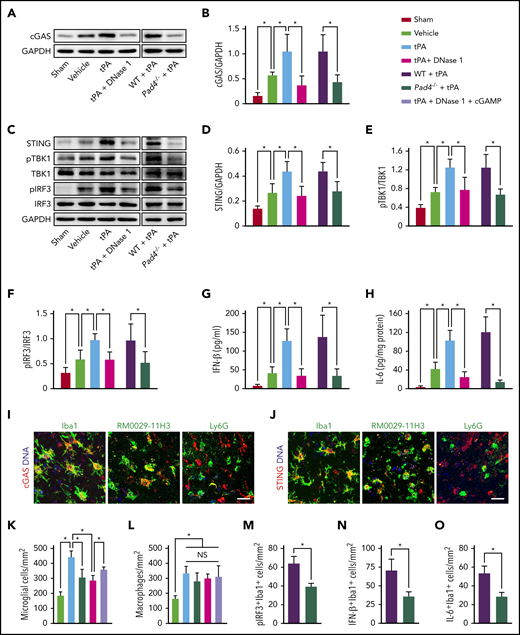
DNase I treatment or PAD4 deficiency inhibits tPA-induced upregulation of cGAS-STING and type 1 IFN signaling. (A-B) Representative immunoblots and quantification of cGAS expression in the ischemic cortex 24 hours after stroke in WT mice treated with vehicle, tPA, or tPA in combination with DNase I and in WT and Pad4−/− mice treated with tPA compared with mice undergoing sham surgery (n = 5). (C) Representative immunoblots for STING, phosphorylated (pTBK1) and total TBK1, and pIRF3 and total IRF3 expression in the ischemic cortex. (D-F) Quantification of band intensities for each group (n = 5). (G-H) Quantification of IFN-β and IL-6 levels by enzyme-linked immunosorbent assay in the ischemic cortex (n = 6). (I-J) Double immunostaining of cGAS and STING with microglial cells (Iba1), macrophages (RM0029-11H3), and neutrophils (Ly6G) in mice subjected to MCAO and tPA treatment. Scale bar, 20 μm. (K-L) Quantification of activated microglia and macrophage infiltration in the ischemic cortex (n = 6). (M-O) Quantification of the numbers of pIRF3+, IFN-β+, and IL-6+ microglial cells (Iba1) in the ischemic cortex (n = 6). Values are means ± standard deviation. *P < .05. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; NS, not significant.
DNase I treatment or PAD4 deficiency inhibits tPA-induced upregulation of cGAS-STING and type 1 IFN signaling. (A-B) Representative immunoblots and quantification of cGAS expression in the ischemic cortex 24 hours after stroke in WT mice treated with vehicle, tPA, or tPA in combination with DNase I and in WT and Pad4−/− mice treated with tPA compared with mice undergoing sham surgery (n = 5). (C) Representative immunoblots for STING, phosphorylated (pTBK1) and total TBK1, and pIRF3 and total IRF3 expression in the ischemic cortex. (D-F) Quantification of band intensities for each group (n = 5). (G-H) Quantification of IFN-β and IL-6 levels by enzyme-linked immunosorbent assay in the ischemic cortex (n = 6). (I-J) Double immunostaining of cGAS and STING with microglial cells (Iba1), macrophages (RM0029-11H3), and neutrophils (Ly6G) in mice subjected to MCAO and tPA treatment. Scale bar, 20 μm. (K-L) Quantification of activated microglia and macrophage infiltration in the ischemic cortex (n = 6). (M-O) Quantification of the numbers of pIRF3+, IFN-β+, and IL-6+ microglial cells (Iba1) in the ischemic cortex (n = 6). Values are means ± standard deviation. *P < .05. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; NS, not significant.
To identify which type of cells expressed cGAS and STING in mice subjected to MCAO and tPA treatment, double immunofluorescence was performed on brain sections. We observed prominent cGAS and STING staining in Iba1+ microglial cells39 and RM0029-11H3+ macrophages40 (Figure 5I-J). By contrast, we found little staining in Ly6G+ neutrophils (Figure 5I-J) and GFAP+ astrocytes (data not shown). Furthermore, we found that treatment with tPA significantly increased the number of activated microglial cells (Figure 5K; supplemental Figure 7A) and the infiltration of macrophages (Figure 5L; supplemental Figure 7B) in the ischemic cortex as reported.41,42 DNase I or PAD4 deficiency similarly reduced the number of activated microglial cells in the ischemic cortex in tPA-treated mice. Moreover, treatment with the cGAS product cGAMP partly reversed the effects of DNase I on tPA-mediated increase in microglial activation. However, neither DNase I nor PAD4 deficiency decreased macrophage infiltration in tPA-treated mice. Because PAD4 deficiency did not significantly affect the infiltration of neutrophils or macrophages in tPA-treated mice, we hypothesize that microglial cells may mediate the changes in inflammation after MCAO that are influenced by PAD4 deficiency. Indeed, deficiency of PAD4 reduced the number of pIRF3+, IFN-β+, and IL-6+ microglial cells in tPA-treated mice with ischemia (Figure 5M-O; supplemental Figure 8A-C).
cGAMP reverses the effects of DNase I on tPA-induced BBB disruption and cerebral hemorrhage
To investigate whether DNase I attenuated tPA-associated BBB disruption and hemorrhage through the cGAS-STING pathway, we infused the cGAS product cGAMP in mice subjected to ischemia and coadministration of DNase I with tPA. The decrease in tPA-induced activation of the cGAS-STING pathway (Figure 6A-D) and induction of IFN-β and IL-6 caused by DNase I (Figure 6E-F) was reversed by cGAMP. Western blot analysis revealed that treatment with cGAMP dramatically increased IgG deposits in capillary-depleted brain tissues in mice treated with tPA plus DNase I (Figure 6G-H). Immunostaining of FITC-dextran with CD31 indicated that infusion of cGAMP abolished the effects of DNase I on tPA-induced vascular leakage of IV injected dextran (Figure 6I-J). Consistent with the increases in BBB permeability, cGAMP reversed the antihemorrhagic effect of DNase I in tPA-treated mice (Figure 6K-L) and worsened neurological deficits (Figure 6M-O) in mice treated with tPA plus DNase I. Together, these data demonstrate that cGAMP blocks the beneficial effects of DNase I on tPA-induced BBB damage and hemorrhage.
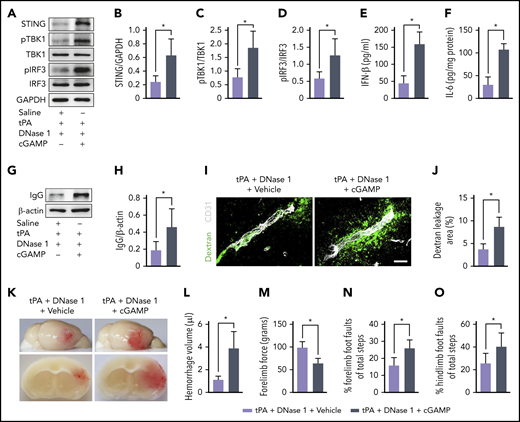
DNase I–mediated cerebrovascular protection and antihemorrhagic effects after ischemic stroke are abolished by cGAMP. (A) Representative immunoblots for STING, pTBK1 and total TBK1, and pIRF3 and total IRF3 expression in the ischemic cortex of tPA-treated mice after treatment with DNase I or DNase I in combination with cGAMP. (B-D) Quantification of band intensities for each group (n = 5). (E-F) Quantification of IFN-β and IL-6 levels by enzyme-linked immunosorbent assay in the ischemic cortex (n = 6). (G-H) Representative immunoblots and quantification of IgG levels in capillary-depleted brain tissue. (I-J) Quantification of extravascular dextran fluorescence in tPA-treated mice after treatment with DNase I or DNase I in combination with cGAMP. Scale bar, 20 μm. At 24 hours after stroke, mice were administered an intravascular injection of 40 KDa of FITC-labeled dextran, and brain sections were stained with CD31. (K) Representative images of the dorsal surface (upper panel) and a coronal section (bottom panel) show cerebral hemorrhage 24 hours after stroke. (L) Quantification of cerebral hemorrhage (n = 8). (M-O) Effects of DNase I or combination of DNase I and cGAMP on forelimb force test and beam walking test 24 hours after stroke in tPA-treated mice (n = 10). Values are means ± standard deviation. *P < .05. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
DNase I–mediated cerebrovascular protection and antihemorrhagic effects after ischemic stroke are abolished by cGAMP. (A) Representative immunoblots for STING, pTBK1 and total TBK1, and pIRF3 and total IRF3 expression in the ischemic cortex of tPA-treated mice after treatment with DNase I or DNase I in combination with cGAMP. (B-D) Quantification of band intensities for each group (n = 5). (E-F) Quantification of IFN-β and IL-6 levels by enzyme-linked immunosorbent assay in the ischemic cortex (n = 6). (G-H) Representative immunoblots and quantification of IgG levels in capillary-depleted brain tissue. (I-J) Quantification of extravascular dextran fluorescence in tPA-treated mice after treatment with DNase I or DNase I in combination with cGAMP. Scale bar, 20 μm. At 24 hours after stroke, mice were administered an intravascular injection of 40 KDa of FITC-labeled dextran, and brain sections were stained with CD31. (K) Representative images of the dorsal surface (upper panel) and a coronal section (bottom panel) show cerebral hemorrhage 24 hours after stroke. (L) Quantification of cerebral hemorrhage (n = 8). (M-O) Effects of DNase I or combination of DNase I and cGAMP on forelimb force test and beam walking test 24 hours after stroke in tPA-treated mice (n = 10). Values are means ± standard deviation. *P < .05. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
cGAS deficiency downregulates tPA-induced STING activation and blocks tPA-mediated brain hemorrhage after stroke
Because cGAMP blocked the effects of DNase I on tPA-mediated activation of the STING pathway, we hypothesized that cGAS is required for STING-dependent type 1 IFN response. Indeed, deficiency of cGAS reduced the levels of STING and STING downstream signaling molecules, including pTBK1 and pIRF3 (Figure 7A-D), and inhibited IFN-β and IL-6 production (Figure 7E-F) in mice subjected to tPA and ischemia. Using multiphoton microscopy of IV injected FITC-dextran, we observed a significant reduction in BBB damage after stroke in tPA-treated cGas−/− mice compared with tPA-treated WT mice (Figure 7G-H). Mice with cGAS deficiency treated with tPA showed a 57% decrease in the leakage of serum IgG after stroke compared with tPA-treated WT mice (Figure 7I-J). We then observed that infusion of tPA in cGas−/− mice with ischemia reduced cerebral hemorrhage by 57% compared with tPA-treated WT mice with ischemia (Figure 7K-L). Furthermore, tPA-treated cGas−/− mice with ischemia displayed increased forelimb force (Figure 7M) and significantly lower foot faults (Figure 7N-O) compared with tPA-treated WT mice with ischemia.
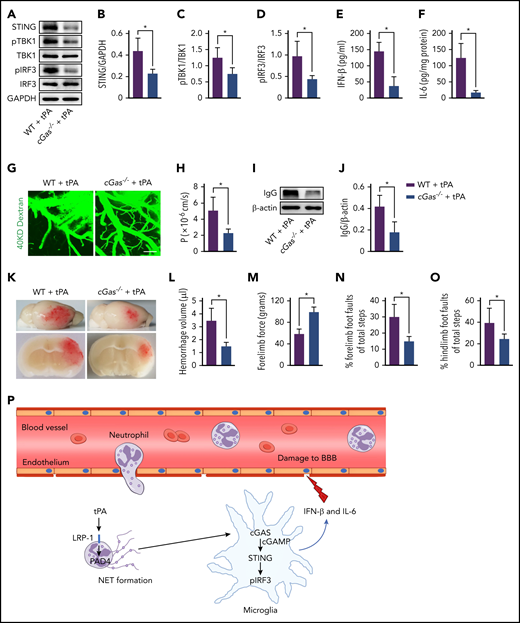
tPA-associated BBB disruption and hemorrhage after ischemic stroke are rescued by loss of cGAS. (A) Representative immunoblots for STING, pTBK1 and total TBK1, and pIRF3 and total IRF3 expression in the ischemic cortex of WT and cGas−/− mice treated with tPA. (B-D) Quantification of band intensities for each group (n = 5). (E-F) Quantification of IFN-β and IL-6 levels by enzyme-linked immunosorbent assay in the ischemic cortex (n = 6). (G-H) Representative in vivo multiphoton microscopic images of IV injected FITC-dextran (molecular weight, 40 KDa) leakage from cortical vessels and quantification of the permeability surface (P) product of FITC-dextran at 24 hours after stroke in WT and cGas−/− mice treated with tPA (n = 6). Scale bar, 100 μm. (I-J) Representative immunoblots and quantification of IgG levels in capillary-depleted brain tissue (n = 5). (K) Representative images of the dorsal surface (upper panel) and a coronal section (bottom panel) show cerebral hemorrhage 24 hours after stroke. (L) Quantification of cerebral hemorrhage (n = 8). (M-O) Effects of cGAS deficiency on forelimb force test and beam walking test 24 hours after stroke in tPA-treated mice (n = 10). (P) Model figure showing how tPA stimulates NET formation and how NETs are sensed by microglia after ischemic stroke. Values are means ± standard deviation. *P < .05. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
tPA-associated BBB disruption and hemorrhage after ischemic stroke are rescued by loss of cGAS. (A) Representative immunoblots for STING, pTBK1 and total TBK1, and pIRF3 and total IRF3 expression in the ischemic cortex of WT and cGas−/− mice treated with tPA. (B-D) Quantification of band intensities for each group (n = 5). (E-F) Quantification of IFN-β and IL-6 levels by enzyme-linked immunosorbent assay in the ischemic cortex (n = 6). (G-H) Representative in vivo multiphoton microscopic images of IV injected FITC-dextran (molecular weight, 40 KDa) leakage from cortical vessels and quantification of the permeability surface (P) product of FITC-dextran at 24 hours after stroke in WT and cGas−/− mice treated with tPA (n = 6). Scale bar, 100 μm. (I-J) Representative immunoblots and quantification of IgG levels in capillary-depleted brain tissue (n = 5). (K) Representative images of the dorsal surface (upper panel) and a coronal section (bottom panel) show cerebral hemorrhage 24 hours after stroke. (L) Quantification of cerebral hemorrhage (n = 8). (M-O) Effects of cGAS deficiency on forelimb force test and beam walking test 24 hours after stroke in tPA-treated mice (n = 10). (P) Model figure showing how tPA stimulates NET formation and how NETs are sensed by microglia after ischemic stroke. Values are means ± standard deviation. *P < .05. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Discussion
For patients with acute ischemic stroke, thrombolytic therapy with tPA remains the only available pharmacological treatment that allows reperfusion of the infarct area.43 However, increased risk of intracerebral hemorrhage limits the use of tPA thrombolysis in ischemic stroke.4,5 Despite extensive efforts, currently there are still no clinically applicable interventions to counteract the adverse effects of tPA on cerebral hemorrhage, because the mechanisms behind it are not fully understood. In this study, we found that tPA treatment in mice with thrombotic stroke increased NET formation. These NETs were associated with increased loss of cerebrovascular integrity, amplified BBB damage, and increased cerebral hemorrhage in tPA-treated mice after stroke, all of which could be attenuated by clearing NETs with DNase I or inhibiting NET production by PAD4 deficiency. We further found that activation of the cGAS-STING pathway and production of IFN-β participated in NET-mediated effects on tPA-associated cerebrovascular complications in stroke.
tPA has been shown to promote neutrophil accumulation in the ischemic brain,44,45 and depletion of neutrophils prevents tPA-induced cerebral hemorrhage.45,46 Here, we found that tPA directly stimulated neutrophils from ischemic mice to release NETs via upregulation of LRP-1 and PAD4. NETs can damage the vascular endothelium and increase vascular permeability by producing cytotoxic proteases, such as histone, elastase, MPO, and proinflammatory mediators.16,47 We found that digestion of NETs with DNase I or inhibition of their formation with PAD4 deficiency reduced the degradation of cerebrovascular integrity and disruption of the BBB. Consistent with evidence showing that disruption of the BBB after tPA treatment is highly predictive for subsequent brain hemorrhage,48,49 we observed that DNase I or PAD4 deficiency similarly decreased tPA-induced hemorrhage, which was associated with significantly improved neurological function in mice with ischemic stroke. This result suggests that limiting excessive NET generation and PAD4 activity is beneficial for tPA therapy in ischemic stroke.
Our results show that tPA treatment significantly amplified the DNA sensor cGAS expression and activation of its adaptor STING in microglial cells and increased the expression of the downstream signals of pTBK1, pIRF3, and IFN-β in the ischemic cortex after stroke. We then found that DNase I suppressed the activation of the cGAS-STING pathway and the production of IFN-β, which was reversed by the cGAS product cGAMP. We also observed that cGAMP partly reversed DNase I–mediated reduction in microglial activation in tPA-treated mice. Moreover, treatment with cGAMP blocked the antihemorrhagic effect of DNase I on tPA-induced bleeding, indicating that the tPA-induced hemorrhage required cGAMP. Therefore, we hypothesize that tPA-mediated cGAS upregulation could lead to deterioration of cerebrovascular integrity after stroke. Indeed, cGAS deficiency normalized tPA-induced disruption of the BBB. Correspondingly, tPA-induced cerebral hemorrhage was inhibited in cGas−/− mice. These results suggest that the antihemorrhagic effect of DNase I was largely mediated by its ability to inhibit cGAS-mediated STING pathway activation.
In summary, although tPA thrombolysis is beneficial for acute ischemic stroke, the increased risk of intracerebral hemorrhage can devastate prognosis. Here, we demonstrate a crucial role for NETs in regulating tPA-induced BBB breakdown and brain hemorrhage. By acting through cGAS-STING pathway–mediated type 1 IFN response, NETs promote tPA-associated vascular complications. We suggest that targeting NETs or cGAS could offer a new approach to improve the safety of tPA thrombolysis for ischemic stroke.
For original data, please contact the corresponding author Bing-Qiao Zhao; e-mail: bingqiaoz@fudan.edu.cn.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
There is a Blood Commentary on this article in this issue.
Acknowledgments
This study was supported by research funding from the National Natural Science Foundation of China (general programs 81671156, 31872777, and 81873744; key program 81530034), National Key Research and Development Program of China, Ministry of Science and Technology of China (2016YFC1300500-501 and 2016YFC1300500-502), and Shanghai Municipal Science and Technology Major Project (2018SHZDZX01) and Zhangjiang Laboratory (ZJLab; Shanghai, China).
Authorship
Contribution: R.W., Y.Z., and Z.L. performed experiments and analyzed and interpreted data; L.C., X.B., L.K., Y.C., X.Y., H.Y., M.-J.S., and Y.H. performed experiments; and R.W., Y.Z., B.-Q.Z., and W.F. designed the study and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Wenying Fan, State Key Laboratory of Medical Neurobiology, Fudan University, 138 Yixueyuan Rd, Shanghai 200032, China; e-mail: wenyingf@fudan.edu.cn; and Bing-Qiao Zhao, State Key Laboratory of Medical Neurobiology, Fudan University, 138 Yixueyuan Rd, Shanghai 200032, China; e-mail: bingqiaoz@fudan.edu.cn.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal