

In this issue of Blood, 1 describe the generation and characterization of new in vivo models of expression of mouse and human interleukin-7 receptor-α (IL7Rα). They also report the impact of IL7Rα expression on the pathogenesis of acute lymphoblastic leukemia and response to therapy. The models fill a critical gap in understanding the biology of lymphoblastic leukemia and IL7Rα. The models can also be used to further study the disease and test therapeutics.
IL7 is a type 1 cytokine and a hematopoietic growth factor secreted by stromal cells in the bone marrow and thymus. It binds to the IL7 receptor, a heterodimer of IL7Rα and the common γ chain receptor (IL2Rγ). The IL7Rα chain also participates in the formation of thymic stromal lymphopoietin receptors. IL7 is important in B- and T-cell development. To this end, the balance of the levels of IL7 and IL7Rα are critical for the homeostasis of the hematopoietic system, and leukemia can develop when this balance goes awry. Indeed, IL7 expression is critical for the growth of leukemia cells.2 Conversely, mice lacking IL7 or IL7Rα have a block in T- and B-cell development, and patients with IL7Rα inactivating mutations present with T-cell−, B-cell+, and natural-killer–cell+ severe combined immunodeficiency.3,4
In 2011, Barata and colleagues reported that 9% of patients with T-cell acute lymphoblastic leukemia (T-ALL), especially those belonging to the TLX3 and HOXA groups, have somatic gain-of-function IL7Ra mutations in their exon 6.5 Similar to T-ALL, patients with B-cell ALL and early T-cell progenitor ALL also present with IL7Ra mutations.6,7 These mutations introduce an unpaired cysteine in the extracellular juxtamembrane-transmembrane region that promotes receptor dimerization and constitutive signaling, independent of IL7 or increased sensitivity for IL7.2,5,7 In vivo modeling of gain-of-function IL7R mutations, by retroviral expression of mutant IL7R in thymocytes on an Arf tumor-suppressor knockout background transplanted into immune-compromised recipients, results in leukemia with full penetrance.8 Past studies have also shown that oncogenes, such as the major T-ALL oncogene NOTCH1, can activate the IL7Ra transcript, and high levels of IL7Rα expression result in increased leukemia-initiating cell activity and leukemia progression.9 The impact of high IL7Rα expression vs mutational activation on leukemogenesis and associated therapeutic implications remains unclear.
In 2 in vivo models, one of the inducible transgenic expressions of the mouse Il7ra and the other of human IL7Ra knocked into the Rosa26 locus, Silva et al demonstrate that ectopic IL7a expression can lead to leukemia with near-complete penetrance, exhibiting characteristics of full-blown leukemia, starting at 20 weeks or earlier. They show that T-cell receptor signaling is not critical for IL7Rα overexpression-induced leukemia, in line with STAT5 transgenic mice, but that elimination of Rag1 expression significantly delays the progression of IL7Rα overexpression-induced leukemia. Shutdown of IL7Rα expression at week 8, with analysis 12 weeks later, showed a reduction in leukemia penetrance with a few mice ultimately developing detectable leukemia, suggesting that early ectopic expression of IL7Rα can commit cells to self-renewal programs.
IL7Ra overexpression results in activation of JAK/STAT, PI3K/AKT, and cell cycle pathways potentially leading to leukemia. Silva et al show that high levels of expression of wild-type IL7Rα in patients with T-ALL correlates with a gene expression signature that significantly overlaps with the signature of IL7Rα mutant cases (see figure). The findings also suggest that subsequent mutations of critical genes (such as Notch1, Atrx, Ptchd,or Idh1) may contribute to disease maintenance in the absence of high Ilr7 levels. Whether IL7Ra overexpression can lead to homodimerization of IL7Rα or heterodimerization with IL2Rγ should be further investigated, especially as IL7Rα associates with JAK1, which signals to STAT1 and 3, whereas the IL7Rα-IL2Rγ heterodimer associates with both JAK1 and JAK3, leading to STAT1, STAT3, and STAT5 activation. Phosphoproteomics analysis with a focus on pathways downstream of IL7Rα would potentially elucidate the mechanisms involved in mutant IL7Rα and high IL7Rα expression.
Silva et al report that IL7Ra-overexpressing cells are sensitive to inhibitors of JAK, STAT, PIM, and PI3K/Akt signaling, as well as acting as a Cdk4/6 inhibitor. These findings provide a rationale for novel therapeutic combinations (see figure). The use of these targeted therapies for downstream pathways is critical, as, surprisingly, high levels of IL7Rα expression may not be necessary after the development of overt leukemia. Thus, the use of IL7Rα antibodies currently being tested for efficacy against leukemia cells may not be efficacious once the leukemia is established.
These mouse models lead to T-ALL development with relatively long latency, and precise comparison of the relative levels of IL7Rα overexpression in the mouse system relative to patients should therefore be performed. This is important from a therapeutic point of view, as high IL7Rα expression may lead to resistance to therapeutics, as has been suggested is the case with glucocorticoids.10
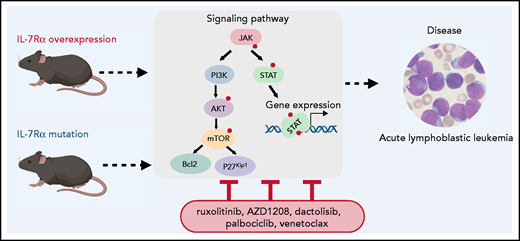
Modeling IL7Ra alterations in leukemia. Expression of mutant ILRα or overexpression of wild-type IL7Rα in thymocytes or hematopoietic progenitors can lead to acute lymphoblastic leukemia with a long latency. These in vivo models are excellent tools for understanding receptor biology and test new therapeutics. Attempts to target downstream signaling pathways leading to leukemia will be facilitated by further characterization of the players (wild-type and mutant receptor) involved in each case. The small red dot represents phosphorylation.
Modeling IL7Ra alterations in leukemia. Expression of mutant ILRα or overexpression of wild-type IL7Rα in thymocytes or hematopoietic progenitors can lead to acute lymphoblastic leukemia with a long latency. These in vivo models are excellent tools for understanding receptor biology and test new therapeutics. Attempts to target downstream signaling pathways leading to leukemia will be facilitated by further characterization of the players (wild-type and mutant receptor) involved in each case. The small red dot represents phosphorylation.
In a significant number of cases, T-ALL presents as refractory to frontline chemotherapy or relapses after initial therapy. Efforts to improve therapy for these patients by targeting oncogenes, such as NOTCH1, have been hampered by toxicity. The present detailed phenotypic characterization of in vivo IL7Ra-overexpressing systems paves the way for future efforts to understand molecular differences between IL7Rα-overexpressing– vs mutation–associated signaling pathways and to test targeted (eg, kinase inhibitors and antiapoptotic drugs) and systemic (chemotherapy) therapeutic approaches for pathways downstream of IL7Rα.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal