

A central challenge in the care of patients with myeloproliferative neoplasms (MPNs) is identifying those individuals at high risk for transformation into acute leukemia, an exacerbation that carries a very poor prognosis.1 In this issue of Blood, 2 describe a novel type of mutation, for which I propose the term “sentinel mutation” (see figure). The acquisition of a “sentinel mutation” drastically increases the likelihood of leukemic transformation, even though in some patients the mutation does not occur in the cells that form the leukemic clone. “Sentinel mutations” function like roses planted in vineyards. Ailing flowers forecast the vines’ impending infection with black rot or downy mildew as the roses are affected first, even if the species that blight the flowers differ slightly from those that spoil the grapes. Similarly, Marcault and colleagues have shown that patients with MPN that acquire mutations in the transcription factor “nuclear factor erythroid 2” (NFE2) carry an increased risk of leukemic transformation, even though in some patients the mutations are not found in the leukemic cells.
The results are remarkable and carry implications beyond the field of MPNs. NFE2 “sentinel mutations” act by a mechanism distinct from that of the 2 previously recognized categories of MPN oncogenic mutations, “driver mutations” and “high-risk mutations.” “Driver mutations” initiate and promote disease development, whereas “high-risk mutations,” which may be incurred in addition, increase the risk of disease exacerbation.3 In most patients, the proportion of cells carrying “driver” and “high-risk” mutations increases during leukemic transformation. In contrast, in some patients, the proportion of cells carrying NFE2 mutations decreased following leukemic transformation, suggesting, as the authors point out, a paracrine mechanism in which the presence of an NFE2 mutation facilitates the acquisition of secondary mutations in other cells of the neoplastic clone. Intriguingly, it is known that in some patients who carry the JAK2V617F driver mutation during the chronic disease phase, the transformed leukemic blasts do not show the JAK2V617F mutation, although they display other genetic abnormalities present during the chronic phase.4,5 These observations demonstrate that leukemic transformation occurred in residual neoplastic cells that had not acquired the JAK2V617F mutation. The novel observations by Marcault et al suggest that paracrine effects on neoplastic subclones in MPN may be more common than previously appreciated (see figure). Moreover, such paracrine stimulation of aggressive subclones may underlie chemotherapy resistance and drive poor prognosis in solid tumors as well.
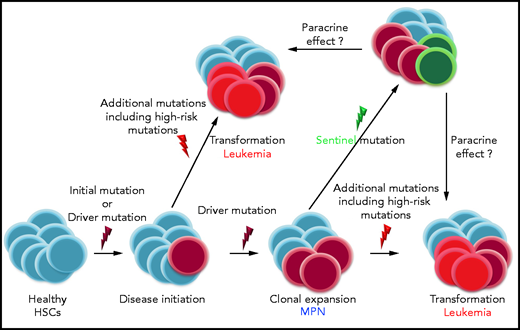
Mechanism of action of the different types of oncogenic mutations in myeloid diseases: “initial mutations” may precede “driver mutations,” which are disease initiating. In MPN, JAK2V617F, CALR, and MPL mutations are recognized as “drivers.” During disease course, additional mutations are incurred; some of these may constitute “high-risk mutations,” for example, ASXL1, EZH2, SRSF2, and IDH1/2, which promote leukemic transformation. Marcault and colleagues describe a novel type of mutation, “sentinel mutations,” the acquisition of which drastically increases the likelihood of leukemic transformation, even though in some patients the mutation does not occur in the cells that form the leukemic clone. The authors suggest that paracrine mechanisms may contribute to leukemic transformation in patients with MPN. It appears that mutations in the same genes, for example, NFE2, can act either as “sentinels” or as “high-risk mutations.” HSCs, hematopoietic stem cells.
Mechanism of action of the different types of oncogenic mutations in myeloid diseases: “initial mutations” may precede “driver mutations,” which are disease initiating. In MPN, JAK2V617F, CALR, and MPL mutations are recognized as “drivers.” During disease course, additional mutations are incurred; some of these may constitute “high-risk mutations,” for example, ASXL1, EZH2, SRSF2, and IDH1/2, which promote leukemic transformation. Marcault and colleagues describe a novel type of mutation, “sentinel mutations,” the acquisition of which drastically increases the likelihood of leukemic transformation, even though in some patients the mutation does not occur in the cells that form the leukemic clone. The authors suggest that paracrine mechanisms may contribute to leukemic transformation in patients with MPN. It appears that mutations in the same genes, for example, NFE2, can act either as “sentinels” or as “high-risk mutations.” HSCs, hematopoietic stem cells.
Mechanistically, we have previously shown in several patients that acquisition of an NFE2 mutation on top of a JAK2V617F mutation provides a proliferative advantage leading to clonal dominance of the doubly mutant clone.6 Likewise, NFE2 mutations are found in de novo acute myeloid leukemia (AML) patients and are sufficient to induce leukemic transformation with the acquisition of AML-specific mutations in murine models.7 It therefore appears that NFE2 mutations can function in different roles: in some patients, they are found in an expanding clone that undergoes leukemic transformation, whereas, in other patients, their proportion decreases as a leukemic clone that carries different mutations outcompetes the once dominant clone. Hence, NFE2 mutations can act either as a “sentinel” or as a “high-risk mutation.” In both cases, their presence indicates a significantly increased risk of leukemic transformation. Of note, we have described 2 different kinds of NFE2 mutations, type I and type II: the former augments NFE2 activity, while the latter acts in a dominant negative manner.7 Further investigations are required to elucidate which signals modulate the effects of altered NFE2 activity, thereby determining the mechanism of action of an NFE2 mutation in a given patient.
For physicians caring for patients with MPN, the results have several direct clinical consequences. One stems from the remarkable size of the observed effect. In multivariate analysis, the presence of an NFE2 mutation carried a hazard ratio of 10.29 for transformation to leukemia and of 8.24 for overall survival, much larger than the hazard ratios of 2.51 and 2.13, respectively, associated with the presence of “high-risk mutations,” which have been validated in many independent cohorts. Therefore, adding NFE2 to the panel of genes routinely sequenced in MPN patients appears prudent. Moreover, as the authors pointed out and Grinfeld et al had previously reported,8 NFE2 mutations are frequently acquired late in the disease course. Consequently, molecular analyses during follow-up, especially when patients lose treatment response, should be considered. Although sampling necessarily introduces a bias in this context, ∼40% of NFE2 mutations were found in patients who had lost response to treatment. More objectively, patients carrying NFE2 mutations incurred significantly lower rates of hematological responses and required significantly more lines of treatment, suggesting that the presence of an NFE2 mutation is a sentinel for a more aggressive disease course in patients with MPN. If validated in independent cohorts, these observations could be implemented in a novel MPN prognostic scoring system.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal