

In this issue of Blood, identify palmitoylation as a novel posttranslational modification that alters mutant FLT3-internal tandem duplication (FLT3-ITD) receptor localization and signaling, and show that modulation of palmitoylation can synergize with FLT3 kinase inhibition to abrogate acute myeloid leukemia (AML) cell growth.1
FLT3 is a class III receptor tyrosine kinase (RTK) that plays an important role in hematopoietic progenitor cell survival and proliferation. Binding of FLT3 ligand leads to FLT3 receptor dimerization, conformational change, autophosphorylation, and stimulation of PI3K/AKT and MAPK/ERK signaling. FLT3 mutations are common in AML, being identified in roughly one-third of patients. The majority of FLT3 mutations involves an ITD in the juxtamembrane domain that leads to constitutive kinase activation. Patients with a FLT3-ITD mutation have a poor prognosis, with an increased tendency to relapse, and higher mortality compared with patients without the mutation. Mutations in the tyrosine kinase domain (TKD) activation loop are seen less commonly, and have less prognostic impact at diagnosis.2
There has been much interest in development of tyrosine kinase inhibitors (TKIs) to target FLT3 for AML treatment. Although first-generation FLT3 inhibitors were limited by lower potency, off- target effects, and rapid development of resistance, the combination of midostaurin with frontline induction chemotherapy nevertheless showed improved survival compared with placebo, and is approved for patients with FLT3-mutated AML. Second-generation TKIs, including gilteritinib, crenolanib, and quizartinib demonstrate higher specificity and potency. Gilteritinib is an oral type I FLT3 inhibitor that binds to the active pocket of the enzyme, which is unaffected by resistance-conferring TKD mutations. Gilteritinib is associated with improved survival in patients with relapsed or refractory FLT3-mutated AML compared with standard chemotherapy and is approved for use in this population. However, activity as a single agent remains limited by drug resistance from secondary NRAS, KRAS, and other MAPK pathway activating mutations or other compensatory signaling mechanisms. Activity may also be reduced by increased levels of FLT3 ligand. These observations suggest that effective FLT3 targeting for AML treatment requires improved understanding of regulation of FLT3 activity and downstream signaling.3
In addition to constitutive kinase activation, FLT3-ITD and TKD mutations also lead to increased receptor localization to the endoplasmic reticulum (ER). There is evidence that cellular compartmentalization alters the downstream signaling pathways activated by FLT3, and that FLT3-ITD localized at the ER aberrantly activates STAT5, whereas targeting of FLT3-ITD to the plasma membrane activates K-RAS, MAPK, and PI3K pathways, with diminished STAT5 activation.4 Therefore, mechanisms underlying FLT3-ITD subcellular localization are of considerable interest. Constitutive tyrosine kinase activity may be a factor because TKI treatment leads to increased localization of FLT3-ITD and FLT3-TKD to the plasma membrane, possibly by inhibiting phospho-dependent interaction with ER proteins.5,6 Posttranslational modification with N-linked branched carbohydrate chains is necessary for receptor maturation and surface expression, and studies suggest that the under-glycosylated immature form predominates in FLT3-ITD expressing cells. Inhibition of FLT3-ITD kinase is reported to promote glycosylation in addition to increasing surface expression. Several reports indicate that glycosylation inhibitors can reduce FLT3-ITD surface expression and signaling.7
The work by Lv et al now identifies lipid modification as a novel mechanism determining FLT3-ITD localization. S-palmitoylation is a reversible posttranslational lipid modification that links the fatty acid palmitate to a cysteine via a thioester bond. Palmitoylation is catalyzed by the ZDHHC family of palmitoyl acyl-transferases and modulates several aspects of protein function, including maturational processing, trafficking, membrane localization, signaling interactions, and stability. Several cancer-associated proteins are known to be palmitoylated, a classic example being the RAS family of small GTPases, where palmitoylation dictates trafficking, membrane localization, and signaling properties.8,9 However, the role of palmitoylation in regulating FLT3-ITD localization and signaling has not been previously shown. Lv et al elegantly demonstrate that S-palmitoylation mediated by ZDHHC6 plays a critical role in determining FLT3-ITD localization and activity (see figure). They show that disruption of palmitoylation promotes trafficking of FLT3-ITD from the ER to the plasma membrane, and leads to activation of AKT and ERK while still maintaining activation of STAT5, and thereby increased FLT3-ITD-mediated leukemic progression. In contrast, palmitoylation did not play a significant role in trafficking of FLT3-WT and TKD mutant proteins to the plasma membrane or their signaling or cellular effects. They further confirmed that FLT3 proteins were palmitoylated, and that ZDHHC6-mediated palmitoylation regulated FLT3-ITD surface expression, signaling and growth in primary human FLT3-ITD+ AML cells.
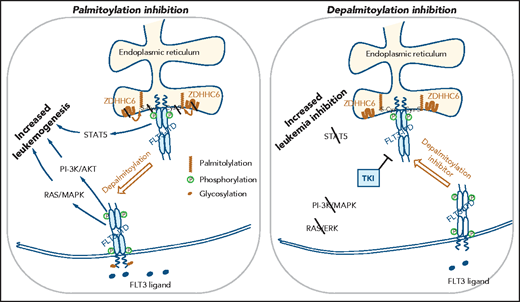
Palmitoylation-mediated alterations in FLT3-ITD localization and signaling. FLT3-ITD localization to the endoplasmic reticulum is enhanced by ZDHHC6-mediated S-palmitoylation. ER-localized FLT3-ITD aberrantly activates cytoplasmic STAT5 that promotes leukemia progression. Inhibition of palmitoylation by mutating the palmitoylated cysteine residue or targeting ZDHHC6 releases FLT3-ITD from the ER to the plasma membrane, where it activates the PI3K/AKT and RAS/MAPK pathways. Combined activation of STAT5, RAS/MAPK, and PI3K/AKT increases AML cell growth. In contrast, treatment with a depalmitoylation inhibitor decreased the cell-surface levels of FLT3-ITD and reduced PI3K/AKT and RAS/MAPK activation and led to enhanced inhibition of AML cell growth in combination with FLT3 TKI treatment.
Palmitoylation-mediated alterations in FLT3-ITD localization and signaling. FLT3-ITD localization to the endoplasmic reticulum is enhanced by ZDHHC6-mediated S-palmitoylation. ER-localized FLT3-ITD aberrantly activates cytoplasmic STAT5 that promotes leukemia progression. Inhibition of palmitoylation by mutating the palmitoylated cysteine residue or targeting ZDHHC6 releases FLT3-ITD from the ER to the plasma membrane, where it activates the PI3K/AKT and RAS/MAPK pathways. Combined activation of STAT5, RAS/MAPK, and PI3K/AKT increases AML cell growth. In contrast, treatment with a depalmitoylation inhibitor decreased the cell-surface levels of FLT3-ITD and reduced PI3K/AKT and RAS/MAPK activation and led to enhanced inhibition of AML cell growth in combination with FLT3 TKI treatment.
It is of note that FLT3-ITD phosphorylation did not affect palmitoylation, and that TKI treatment further increased the surface levels of a palmitoylation-deficient ITD mutant, suggesting that palmitoylation and phosphorylation are separate mechanisms regulating FLT3-ITD intracellular localization. The relationship of palmitoylation to receptor glycosylation and maturation was not evaluated, and requires further study. Palmitoylation-deficient FLT3-ITD mutants retained sensitivity to gilteritinib. Importantly, pharmacological inhibition of FLT3-ITD depalmitoylation using a pan-depalmitoylase inhibitor significantly reduced FLT3-ITD surface expression, inhibited AKT and ERK signaling, and reduced cell growth. The depalmitoylase inhibitor synergized with Gilteritinib in inhibiting FLT3-ITD surface localization, AKT and ERK signaling, and abrogating growth of primary FLT3-ITD+ AML cells. These observations provide new insights into the role of lipid modifications in compartmentalization of FLT3-ITD signaling in AML. Importantly, they indicate that targeting of depalmitoylation could be a potential therapeutic strategy for FLT3-ITD+ leukemias and support further exploration and development of clinically applicable inhibitors of depalmitoylation. Because resistance to gilteritinib has been associated with reactivation of RAS/MAPK pathway, it will be of interest to determine whether depalmitoylation inhibitors provide additional benefit in FLT3-ITD+ AML through inhibition of RAS/MAPK signaling.3
The implications of these studies extend beyond FLT3-ITD AML because subcellular localization is a general mechanism that affects activation of RTKs and their downstream pathways.7 Abnormal maturation and trafficking have also been observed for other oncogenic RTKs, which in addition to being aberrantly active in different cellular compartments, can also generate different signaling outputs depending on localization. The role of palmitoylation in localization, signaling, and transforming activity of other RTKs will doubtless be the subject of future studies.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal