

In this issue of Blood, used single-cell RNA sequencing (scRNA-seq) to identify a unique and dominant role for STAT5B in self-renewal of hematopoietic stem cells (HSCs) and leukemia stem cells (LSCs). Moreover, they found that the cell surface marker CD9 is an important STAT5B target gene, implicating CD9 as a novel therapeutic target for STAT5-driven leukemia.1
STAT5A and STAT5B are 2 of the 7 members of the STAT family, and both are activated by a broad spectrum of cytokines and growth factors.2 STAT5A and STAT5B are 95% identical at the amino acid sequence level. They are commonly believed to have redundant roles in the hematopoietic system, because both are required for lymphocyte development and T-cell proliferation and function.3-5 Both STAT5A and STAT5B are important for the repopulating potential of HSCs6; However, it is not known whether they play distinct roles in HSCs or uniquely activate target genes in rare HSC populations. Kollmann et al identified the unexpected predominant role of STAT5B in controlling the expression of genes associated with quiescence and self-renewal of HSCs and LSCs (see figure).
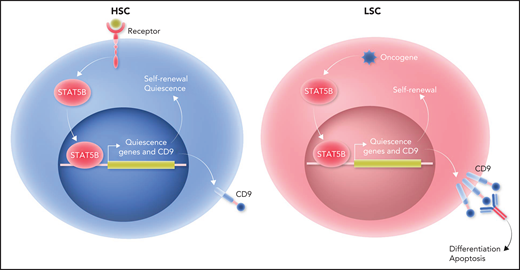
Only STAT5B plays a distinct and dominant role in self-renewal of HSCs and LSCs. Left panel: receptor engagement predominantly activates STAT5B in HSCs. STAT5B, in turn, selectively activates a set of “quiescence” genes that drive self-renewal and quiescence of HSCs. One of the STAT5B target genes is CD9. Right panel: the dominant role of STAT5B extends to LSCs. Oncogene stimulation predominantly activates STAT5B in LSCs. Subsequently, STAT5B selectively activates a set of “quiescence” genes that drive self-renewal of LSCs. CD9 expression levels are higher in LSCs than in HSCs. Blocking CD9 by antibodies can induce differentiation and apoptosis in STAT5B-driven LSCs, leading to their eradication. Professional illustration by Somersault18:24.
Only STAT5B plays a distinct and dominant role in self-renewal of HSCs and LSCs. Left panel: receptor engagement predominantly activates STAT5B in HSCs. STAT5B, in turn, selectively activates a set of “quiescence” genes that drive self-renewal and quiescence of HSCs. One of the STAT5B target genes is CD9. Right panel: the dominant role of STAT5B extends to LSCs. Oncogene stimulation predominantly activates STAT5B in LSCs. Subsequently, STAT5B selectively activates a set of “quiescence” genes that drive self-renewal of LSCs. CD9 expression levels are higher in LSCs than in HSCs. Blocking CD9 by antibodies can induce differentiation and apoptosis in STAT5B-driven LSCs, leading to their eradication. Professional illustration by Somersault18:24.
To determine the unique functions of STAT5A and STAT5B in HSCs, Kollmann et al used scRNA-seq to reveal that 2 cell clusters with gene signatures representative of dormant HSCs were decreased in Stat5b-deficient mice relative to wild-type or Stat5a-deficient mice. Flow cytometry analysis confirmed the reduction of dormant HSCs in Stat5b-deficient mice. Cell cycle analysis, as well as single-cell culture and replating assays, demonstrated that Stat5b-deficient HSCs exhibited a drastically reduced self-renewal potential. Overexpression of Stat5b, but not Stat5a, enhanced HSC growth. In addition, serial bone marrow transplantation assays revealed that the repopulating ability of Stat5b-deficient HSCs decreased gradually, which confirmed that STAT5B intrinsically drives HSC self-renewal. These findings demonstrate that STAT5B, but not STAT5A, plays a distinct and dominant role in self-renewal of normal HSCs (see figure). LSCs, which initiate and maintain leukemia, share many characteristics with HSCs. Therefore, Kollmann et al further investigated whether the dominant role of STAT5B extends to LSCs. In vitro serial plating and in vivo serial transplantation experiments showed that BCR/ABLp210-induced proliferation and transformation of HSC-containing Lin−Sca-1−cKit+ (LSK) cells depended on STAT5B but not STAT5A. Furthermore, the oncogenes JAK2V617F and FLT3-ITD predominantly activated STAT5B in LSK cells. Human acute myeloid leukemia (AML) and chronic myelogenous leukemia cell lines also had a higher level of STAT5B activation relative to STAT5A activation. These data demonstrate that STAT5B plays the dominant role in driving LSC self-renewal (see figure).
STAT5B-regulated genes might be therapeutic targets for specifically eradicating LSCs. Kollmann et al performed differential gene-expression analysis of the most dormant and other HSC subpopulations in wild-type, Stat5a-deficient, and Stat5b-deficient mice and discovered that 35 genes were specifically regulated by STAT5B in HSCs. Interestingly, among the STAT5B target genes identified, CD9 was the only one that correlated negatively with overall survival of human patients with STAT5-driven, but not non–STAT5-driven, leukemia. Relative to patients with AML with low CD9 expression, those with high CD9 expression had higher levels of expression of STAT5 target genes. Conversely, CD9 expression was reduced in Stat5b-deficient, but not Stat5a-deficient, LSK cells. Moreover, chromatin immunoprecipitation quantitative polymerase chain reaction demonstrated that STAT5B binds directly to the CD9 promoter. Hyperactive STAT5B, but not STAT5A, in LSK cells or activated STAT5B by oncogenes in LSK cell lines was associated with enhanced CD9 expression. Thus, STAT5B directly drives the expression of CD9. Importantly, blocking CD9 by antibodies could induce differentiation and apoptosis of STAT5B-driven LSCs in mouse models and human patient samples. Taken together, these findings suggest that targeting CD9 may enable eradication of LSCs while largely sparing HSCs and implicate the STAT5B target gene CD9 as a new therapeutic target for STAT5-driven leukemia (see figure).
Why is STAT5B the dominant twin for self-renewal of HSCs and LSCs? Previous studies demonstrated that STAT5A and STAT5B can have distinct functions, despite their high degree of sequence homology.2 STAT5A uniquely regulates prolactin-mediated mammary gland function, whereas STAT5B is critical for growth hormone–regulated functions.7 STAT5B also plays a dominant role in the development and function of lymphocytes.8 Unique functions of STAT5A vs STAT5B correlate with tissue-specific differences in their relative levels of expression.7,8 Intriguingly, Kollmann et al found that STAT5A and STAT5B were expressed at similar levels in HSCs and that stimulation of HSCs with growth factors mostly activated STAT5B, and oncogenes preferentially activated STAT5B in hematopoietic malignancies.1 STAT5A and STAT5B differ primarily in their Src-homology 2 (SH2) domains, which bind specific phosphotyrosine-containing motifs in the receptors with which they associate.2 However, SH2 domain dissimilarities between STAT5A and STAT5B cannot account for the selective activation of STAT5B in HSCs because growth factor stimulation activates STAT5A and STAT5B in megakaryocytes.1 Differences in the nature/extent of posttranslational modifications of STAT5A and STAT5B and/or the presence of specific regulators in HSCs and LSCs could impact the receptor interactions or activation states of STAT5A or STAT5B. These findings suggest that selective activation of STAT5B underlies its unique and dominant role in self-renewal of HSCs and LSCs. Proteomic analysis might help to further elucidate the molecular mechanism underlying selective activation of STAT5B in these cells.
The different transcriptional signatures of STAT5A and STAT5B further complicate understanding of the dominant role of STAT5B in HSCs and LSCs. Selective activation of “quiescence” genes by STAT5B could explain why STAT5B, but not STAT5A, is able to drive self-renewal and quiescence. Dissimilarities located in the transactivation domains of STAT5A and STAT5B might contribute to the distinct transcriptional signatures of STAT5B in HSCs and LSCs.2 Thus, the STAT5B transactivation domain might specifically recruit a unique partner in HSCs and LSCs to facilitate its promoter binding and/or transcriptional activation of “quiescence” genes, which may also be influenced by the distinct epigenetic landscape of HSCs and LSCs. Future proteomic and epigenetic analyses of HSCs and LSCs might help to reveal the molecular mechanism by which STAT5B, but not STAT5A, activates “quiescence” genes in these cells.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal