


Abstract
Autoimmune conditions can occur in a temporary relationship with any malignant lymphoma. In many instances, treatment at diagnosis is not required, but symptomatic autoimmune conditions represent an indication for treatment, particularly in chronic lymphoproliferative diseases. Treatment is selected depending on the predominant condition: autoimmune disease (immunosuppression) or lymphoma (antilymphoma therapy). Steroids and anti-CD20 antibodies are effective against both conditions and may suppress the autoimmune complication for a prolonged period. The efficacy of B-cell receptor inhibitors has provided us with novel insights into the pathophysiology of antibody-producing B cells. Screening for underlying autoimmune conditions is part of the lymphoma workup, because other drugs, such as immunomodulators and checkpoint inhibitors, should be avoided or used with caution. In this article, we discuss diagnostic challenges and treatment approaches for different situations involving lymphomas and autoimmune cytopenias.
Introduction and background
The association between lymphoid neoplasms and autoimmune diseases has long been noted. Definitive evidence linking the causality of the diseases is rare. However, there is compelling evidence of their co-occurrence.1,2 Autoimmune diseases occur with a prevalence of ∼10% in lymphoid neoplasms.3 However, the frequency varies among entities, with a high prevalence in certain lymphomas (from 7.4% in Hodgkin lymphoma to 18% in marginal zone lymphoma).4,5 Most autoimmune conditions (AICs) are associated with B-cell lymphomas and are much less frequent with T-cell non-Hodgkin lymphomas (NHLs).
Autoimmune complications in lymphoproliferative diseases frequently affect the hemopoietic system, but may have rheumatologic, endocrinologic, neurologic, or other manifestations.6 In this article we focus on the management of autoimmune cytopenias illustrated by practical cases.
Autoimmune cytopenias can occur at any time during the course of a lymphoproliferative disease: (1) before lymphoma diagnosis, (2) at diagnosis of lymphoma, and (3) after diagnosis or treatment of lymphoma.7 In some cases, the autoimmune phenomenon may even be related to antilymphoma therapy.7-9 The pathophysiology of autoimmune cytopenias in relation to the time of occurrence, as well as to their association with predisposing diseases (eg, rheumatic diseases) or treatments may be very different.
Among autoimmune cytopenias, autoimmune hemolytic anemia (AIHA) and immune thrombocytopenia (ITP) are the most frequent, but other conditions occur, such as immune-mediated neutropenia or pure red cell aplasia (PRCA). The diagnosis of autoimmune cytopenia is usually made according to the criteria of the respective disease. In some cases, antibodies are detected without symptomatic disease. B-cell–mediated (antibody-dependent) AICs are predominant, but T cells may also play an important role.4 The antibodies involved may be monoclonal (eg, IgMκ in cold agglutinin disease [CAD]) or polyclonal (eg, warm antibodies in warm AIHA [wAIHA]).
Clinical management consists of appropriate diagnostic workup, but the most important decision to make is whether treatment is needed, and, if so, whether it should be directed against the AIC, the lymphoma, or both. In this report, we describe several typical scenarios, with practical solutions and recommendations.
AIHA
AIHA may occur shortly before diagnosis of lymphoma or may be detected during the lymphoma workup. AIHA is best characterized in chronic lymphocytic leukemia (CLL), but is found as a complication in many lymphoma entities and in solid tumors.10-17 Severe AIHA may lead to initiation of antilymphoma treatment, but in many cases, immunosuppression stabilizes the condition for further watchful waiting (Figure 1A-C). The International Workshop on Chronic Lymphocytic Leukemia Guidelines, for instance, explicitly state that AICs mimicking a CLL Binet C stage can be treated with immunosuppressive therapy.18 A direct antiglobulin test (DAT) is part of the CLL screening program, and the result is positive in up to 14% of patients at diagnosis.11,18 Reticulocytes are elevated, and biochemical signs of hemolysis, such as elevated bilirubin and lactate dehydrogenase (LDH), are usually present. Other features pointing to AIHA are morphologic abnormalities of the blood smear (polychromasia and spherocytosis). The prevalence of AIHA in CLL is ∼2.9% in stable Binet disease stage A compared with 10.5% in stages B and C.19 Clinically, a discrepancy between a normal platelet count and anemia with high reticulocytes should lead to the suspicion of AIHA. The anti–red cell autoantibody involved in CLL is predominantly of the IgG warm type, but cold agglutinins of the IgM type have been observed.20 If the diagnosis of warm AIHA is established, and the hemoglobin is <10 g/dL or the patient is symptomatic, prednisone at a dose of 1 mg/kg is usually effective and should be continued according to AIHA treatment recommendations (Table 1).21-23 Rituximab (4 times weekly at 375 mg/m2) is recommended if steroids fail. Rituximab responses are high (71% of cases) but are not always long-lasting.12,24 Because of its antilymphoma activity, it can also be used in first-line treatment (eg, if steroid treatment is contraindicated). Rituximab is also the treatment of choice if cold agglutinins are present (Table 1).21
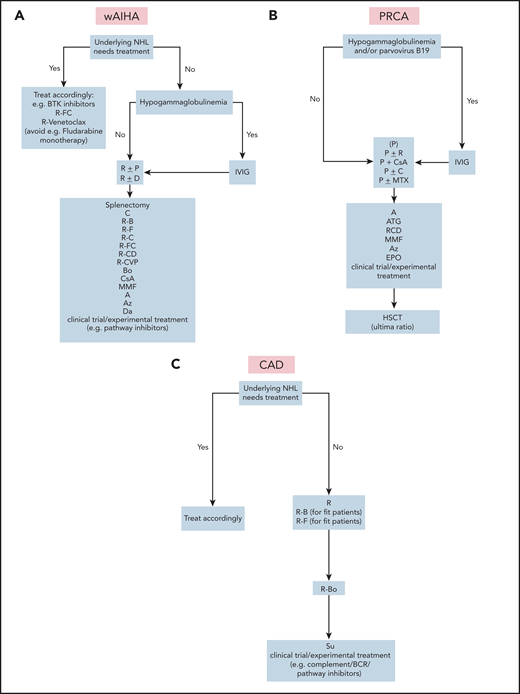
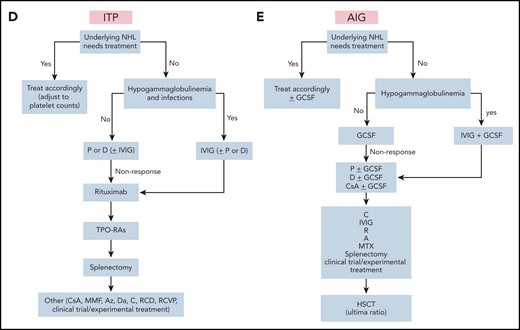
Treatment algorithms for autoimmune cytopenias in lymphoma. (A) wAIHA. Supportive care: red blood cell transfusions, if hemoglobin level is <8 mg/dL and/or there is symptomatic anemia. Lower hemoglobin levels may be tolerated in well-adapted and/or young adults; higher hemoglobin levels are needed in cases of additional heart diseases or symptomatic anemia in elderly patients. Observe serum ferritin levels to avoid iron overload. Start iron chelation therapy if necessary, add folic acid, osteoporosis inhibitor, and thrombosis prophylaxis. (B) PRCA. Supportive care: as described in panel A, except that folic acid or osteoporosis treatments or thrombosis prophylaxis are not used. (C) CAD. Supportive care: as in panel A, except that folic acid or osteoporosis treatment or thrombosis prophylaxis are not used. (D) ITP. Supportive care: platelet transfusions only for clinically significant bleeding or with surgical intervention. (E) AIG. Supportive care: consider granulocyte transfusions to treat severe sepsis. Detailed regimens are given in Table 1.
Treatment algorithms for autoimmune cytopenias in lymphoma. (A) wAIHA. Supportive care: red blood cell transfusions, if hemoglobin level is <8 mg/dL and/or there is symptomatic anemia. Lower hemoglobin levels may be tolerated in well-adapted and/or young adults; higher hemoglobin levels are needed in cases of additional heart diseases or symptomatic anemia in elderly patients. Observe serum ferritin levels to avoid iron overload. Start iron chelation therapy if necessary, add folic acid, osteoporosis inhibitor, and thrombosis prophylaxis. (B) PRCA. Supportive care: as described in panel A, except that folic acid or osteoporosis treatments or thrombosis prophylaxis are not used. (C) CAD. Supportive care: as in panel A, except that folic acid or osteoporosis treatment or thrombosis prophylaxis are not used. (D) ITP. Supportive care: platelet transfusions only for clinically significant bleeding or with surgical intervention. (E) AIG. Supportive care: consider granulocyte transfusions to treat severe sepsis. Detailed regimens are given in Table 1.
Regimens used in the treatment of autoimmune cytopenias in lymphoma
| Abbreviation | Therapy | Dose | References |
|---|---|---|---|
| A | Alemtuzumab | 3-mg test dose, then 10-30 mg/wk for 4-6 wk | 43 |
| ATG | Anti-thymocyte globulin | 10-20 mg/kg for 10 d | 45 |
| Az | Azathioprine | 0.5 mg/kg initial up to 2-2.5 mg/kg day (max, 150 mg/d) | 21 |
| B | Bendamustine | 70 or 90 or 100 mg/every morning on days 1-2 every 4 wk | 28, 55, 59 |
| Bo | Bortezomib | 1.3 mg/wk for 1-4 wk | 21, 55, 61 |
| C | Cyclophosphamide | 1-2 mg/kg per day or 50-150 mg/d orally or 300-1000 mg IV on day 1 every 4 wk (eg, 750 mg per day) | 13, 22, 26, 30-31 |
| CsA | Cyclosporin A | 3-5 mg/kg per day | 19, 21 |
| ct/et | Clinical trials/experimental therapies | eg, ibrutinib 420 mg/d or venetoclax 400 mg/d after ramp-up weeks | 8, 20-21, 34-35, 41, 49, 55, 65 |
| D | Dexamethasone | 40 mg for 4 d (monotherapy) or 12 mg IV on days 1-2 and orally on days 3-7 (eg, combined with rituximab and cyclophosphamide) | 11-13, 21-22, 30-31, 69 |
| Da | Danazol | 200 mg 3-4 times per day | 21-22 |
| EPO | Epoetin α | 5 000-40 000 U/wk | 44-45 |
| F | Fludarabine | 40 mg every morning on days 1-5 or 25 mg every morning on days 1-3 | 22, 26, 55, 58 |
| GCSF | Granulocyte colony stimulating factor | 1-3 μg/kg per day | 91 |
| HSCT | Hematopoietic stem cell transplantation | — | — |
| IVIG | Intravenous immune globulin | 1-2 g/kg per day over 1-2 d (PRCA: 5 d) | 12-13, 19-20, |
| MMF | Mycophenolate mofetil | 600 mg/d, 2 times per day | 19, 21 |
| MTX | Methotrexate | 10 mg every morning weekly | 77 |
| P | Prednisone | 0.5-2 mg/kg per day for 4 wk, taper over 1-2 mo | 11-13, 21-22, 69 |
| R | Rituximab | 375 mg/every morning weekly for 4 wk (or 100 mg weekly or 1000 mg on the days 1 and 15) | 12-13, 19, 21-22, 28, 30-31, 55-56, 58-59, 69 |
| Su | Sutimlimab | 10 mg/kg test dose, 60 mg/kg 4 times per week starting 1-4 d after test dose | 55, 65 |
| TPO-RAs | Thrombopoietin receptor agonists | Eltrombopag 50-150 mg/d, romiplostim 1-10 μg/kg per week | 69-70, 92 |
| V | Vincristine | 1 mg/wk for 4-6 wk | 19, 32 |
| Abbreviation | Therapy | Dose | References |
|---|---|---|---|
| A | Alemtuzumab | 3-mg test dose, then 10-30 mg/wk for 4-6 wk | 43 |
| ATG | Anti-thymocyte globulin | 10-20 mg/kg for 10 d | 45 |
| Az | Azathioprine | 0.5 mg/kg initial up to 2-2.5 mg/kg day (max, 150 mg/d) | 21 |
| B | Bendamustine | 70 or 90 or 100 mg/every morning on days 1-2 every 4 wk | 28, 55, 59 |
| Bo | Bortezomib | 1.3 mg/wk for 1-4 wk | 21, 55, 61 |
| C | Cyclophosphamide | 1-2 mg/kg per day or 50-150 mg/d orally or 300-1000 mg IV on day 1 every 4 wk (eg, 750 mg per day) | 13, 22, 26, 30-31 |
| CsA | Cyclosporin A | 3-5 mg/kg per day | 19, 21 |
| ct/et | Clinical trials/experimental therapies | eg, ibrutinib 420 mg/d or venetoclax 400 mg/d after ramp-up weeks | 8, 20-21, 34-35, 41, 49, 55, 65 |
| D | Dexamethasone | 40 mg for 4 d (monotherapy) or 12 mg IV on days 1-2 and orally on days 3-7 (eg, combined with rituximab and cyclophosphamide) | 11-13, 21-22, 30-31, 69 |
| Da | Danazol | 200 mg 3-4 times per day | 21-22 |
| EPO | Epoetin α | 5 000-40 000 U/wk | 44-45 |
| F | Fludarabine | 40 mg every morning on days 1-5 or 25 mg every morning on days 1-3 | 22, 26, 55, 58 |
| GCSF | Granulocyte colony stimulating factor | 1-3 μg/kg per day | 91 |
| HSCT | Hematopoietic stem cell transplantation | — | — |
| IVIG | Intravenous immune globulin | 1-2 g/kg per day over 1-2 d (PRCA: 5 d) | 12-13, 19-20, |
| MMF | Mycophenolate mofetil | 600 mg/d, 2 times per day | 19, 21 |
| MTX | Methotrexate | 10 mg every morning weekly | 77 |
| P | Prednisone | 0.5-2 mg/kg per day for 4 wk, taper over 1-2 mo | 11-13, 21-22, 69 |
| R | Rituximab | 375 mg/every morning weekly for 4 wk (or 100 mg weekly or 1000 mg on the days 1 and 15) | 12-13, 19, 21-22, 28, 30-31, 55-56, 58-59, 69 |
| Su | Sutimlimab | 10 mg/kg test dose, 60 mg/kg 4 times per week starting 1-4 d after test dose | 55, 65 |
| TPO-RAs | Thrombopoietin receptor agonists | Eltrombopag 50-150 mg/d, romiplostim 1-10 μg/kg per week | 69-70, 92 |
| V | Vincristine | 1 mg/wk for 4-6 wk | 19, 32 |
Patient 1
A 67-year-old man with typical CLL experienced a sudden decrease in hemoglobin from 12.9 to 9.3 g/dL, with an increase in reticulocytes to 163 × 109/L and mean corpuscular volume of 110 fL, whereas platelets were still normal. Haptoglobin was reduced (<25 mg/dL) with slightly elevated indirect bilirubin and LDH. The DAT result was positive for anti-IgG1 but not for complement C3d, consistent with wAIHA. Standard treatment with prednisolone at 1 mg/kg body weight was initiated. The anemia responded within 1 month (Hb, 11.0 g/dL; reticulocytes, 74 × 109/L), prednisolone was slowly tapered, and hemoglobin increased to 13.5 g/dL in September 2012, when the steroids were discontinued. The DAT result was still positive, which is a common finding. However, 4 months later, the AIHA reoccurred, with a hemoglobin of 8.7 g/dL. The patient was then treated with 4 weekly doses of rituximab 375 mg/m2 and had a complete response (CR) of wAIHA with a persistent DAT positivity for IgG. Eighteen months later, the CLL had progressed to Binet stage C with thrombocytopenia, without signs of ITP in the blood or bone marrow, but again accompanied by AIHA. A bone marrow biopsy specimen showed heavy infiltration with CLL and a reduced number of megakaryocytes. The patient was treated with 6 cycles of rituximab and bendamustine (absence of TP53 abnormalities and mutated IgHV status) and hematological CR as well as a CR of AIHA was achieved. At this writing, the patients is 78 years of age and is in ongoing CR. This case shows the spectrum of treatment options ranging from immunosuppression to antilymphoma treatment.
Treatment of AIHA and/or lymphoma with chemoimmunotherapy or novel agents
wAIHA
Patients experiencing progression of underlying lymphoma (in this case, CLL) may be better treated with chemoimmunotherapy (Figure 1A). Regimens containing anti-CD20 antibodies are effective against both the AIC and the lymphoma. However, fludarabine (particularly without rituximab) should be avoided because of its potential to cause AIHA by itself.25-27 Rituximab+bendamustine has shown excellent effects, with response rates of 81% for AIHA and 77% for CLL (Table 1).28 These results outweigh the few cases of complicating AIHA after bendamustine therapy.29 Treatment with rituximab, cyclophosphamide, and dexamethasone is another good choice, particularly in CLL or indolent lymphomas. 30,31 Some patients may even convert to Coomb’s negativity. The treatment is effective for other autoimmune cytopenias in CLL. A combination of rituximab with cyclophosphamide, vincristine, and prednisone has also been used.32 In contrast to primary wAIHA, splenectomy is not a major option in lymphoma-associated AIHA because of an increased risk of infection.33
The Bruton’s tyrosine kinase (BTK) inhibitor ibrutinib is the preferred option in patients who do not respond to chemoimmunotherapy or those with TP53 abnormalities. Ibrutinib induces long-term responses in AIHA and CLL.34-38 A low rate of treatment-emergent AICs has been reported.9,39 The Bcl-2 inhibitor venetoclax seems to be effective against AIHA as well, although few cases have been published so far.40,41 Novel anti-CD20 antibodies such as ofatumumab or obinutuzumab should have activity that is at least similar to that of rituximab, but published data are rare.42
CLL guidelines now recommend the use of BTK or Bcl2 inhibitors in most cases. The low rate of autoimmune complications associated with these therapies is outweighed by the antilymphoma effect. With large data sets still missing, we prefer sequential therapy or simultaneous administration of an anti-CD20 antibody in patients with AIHA in need of antilymphoma therapy with ibrutinib or venetoclax.23
The following case describes treatment of a patient with CLL with del(17p) with ibrutinib accompanied by an unusual complication.
Patient 2
A 35-year-old woman was diagnosed with CLL with a TP53 mutation and deletion (17p). Her white blood cell (WBC) count was 93 × 109/L, with a platelet count of 239 × 109/L, and severe anemia, with a hemoglobin level of 6.5 g/dL. Low haptoglobin and elevated LDH levels and total bilirubin were noted, but the DAT result was negative (as it is in 5% of AIHA cases).21 Interestingly, the patient had a very low reticulocyte count (0.007 × 1012/L; normal value, 0.027 to 0.116 × 1012/L). A bone marrow examination revealed typical CLL but suppressed erythropoiesis. A diagnosis of anemia related to CLL was made, indicating Binet stage C or CLL with PRCA (based on low reticulocyte counts and diminished erythropoiesis in the bone marrow) with DAT-negative AIHA. The patient was dependent on blood transfusions. CLL-specific treatment with ibrutinib was initiated and resulted in normalization of WBCs, but without hemoglobin response (6 g/dL). We reasoned that the AIC was predominant in this case. Therefore, ibrutinib was discontinued, and the patient was treated with prednisone at 1 mg/kg, followed by 4 weekly doses of rituximab. Hemoglobin increased to normal levels (14.9 mg/dL). Ibrutinib was reinstated, and, at this writing, the patient is still in CR of CLL and PRCA.
This case shows that AICs can dramatically influence the course of disease, with immediate need for treatment. The diagnosis of autoimmune anemia is not always easy to make. In this case, the diagnosis of AIHA could never be clearly established because of the absence of a positive DAT result, whereas the patient had low haptoglobin and elevated LDH and bilirubin. The DAT negativity may have been a consequence of low-affinity autoantibodies or warm IgM or IgA antibodies. The key finding in this case was the suppressed erythropoiesis in the bone marrow, together with the almost absent reticulocyte count. However, PRCA is a finding encountered in lymphoid neoplasms and should be differentiated from, for example, parvovirus infection. In this case, we had no information about parvovirus or T-cell receptor rearrangement. The latter also indicates that PRCA lymphoma is, at least in part, caused by suppression of erythropoiesis by T cells. PRCA in lymphomas may occur alone or in conjunction with AIHA (Figure 1B).
As discussed earlier, ibrutinib is effective against CLL-associated AIHA in most cases. However, in this case, the AIC did not respond to the BTK inhibitor, possibly because of a T-cell–mediated mechanism. It responded only to adequate doses of steroids (and rituximab) given over a lengthy period, as also recommended for AIHA.22 It is important to use a dose of 1 mg/kg and not to taper the treatment too early. Subsequent treatment with ibrutinib in our patient was administered without problems and led to a CR of CLL. It is important to note that combined treatment with ibrutinib and steroids can lead to serious complications, specifically opportunistic infections (eg, invasive fungal infections).43 These complications were the major reason that ibrutinib and prednisone were given sequentially to patient 2.
Cold agglutinin disease
The pathophysiology of cold agglutinin disease (CAD) is based on monoclonal IgM antibodies that trigger complement-mediated red cell lysis,48-53 which causes predominantly extravascular hemolysis in the liver. Effective removal of the B-cell clone and complement inhibition are therefore the therapeutic goals.54,55 For these reasons, CAD usually does not respond to steroids or splenectomy. Rituximab or rituximab-containing regimens are very effective as first-line treatments (Table 1; Figure 1C). Rituximab monotherapy, 375 mg/m2 for 4 weeks at 7-day intervals, produces response rates of ∼50%, but with few CRs.21,56,57 The median duration of response is less than 1 year, but repeated courses of rituximab are often effective in relapsed disease. A good option for primary CAD, particularly cases of accompanying indolent lymphomas, are combinations of rituximab with fludarabine or bendamustine.58,59 We prefer bendamustine+rituximab for its higher efficacy and rare induction of hemolytic events. Bortezomib is another effective drug for treating lymphomas.60,61 Ibrutinib is effective in Waldenström’s disease and can be tried as a monotherapy.62,63 In case of AICs, a CD20 monoclonal antibody may increase efficacy.64
The other option for CAD is complement inhibition. The anti-C1s antibody sutimlimab effectively increased hemoglobin levels rapidly by more than 2 g/dL in 7 of 10 patients, with a median best response of 3.9 g/dL.65 Inhibition of the complement cascade offers short-term relief, but does not target the underlying clonal B-cell disease.66 However, it would be a good option for patients who do not respond to rituximab or as a bridging therapy. Sutimlimab can also be used in sequence with ibrutinib in patients with lymphoplasmacytic lymphoma with an MYD88 mutation.65,67 Clinical studies with other B-cell–targeting agents such as PI3 kinase or SYK inhibitors are ongoing.
Patient 3
A 63-year-old man, who presented with severe autoimmune hemolytic anemia after a hip replacement, was prepared for hip surgery in January 2014. Postoperative hemoglobin level was 7.8 g/dL with elevated reticulocyte count at 183 × 109/L, haptoglobin below the detection limit, and bilirubin at 2.8 mg/dL. LDH was elevated, and a DAT result was positive for complement anti-C3d antibodies. The cold agglutinin titer was 64. The patient had an IgMκ monoclonal protein, however, with normal absolute IgM serum levels (209 mg/dL). A diagnosis of cold AIHA was made, and, on further hematologic workup, a slightly enlarged spleen was observed, as well as an Igκ and IgHV rearrangement in the peripheral blood by polymerase chain reaction. The bone marrow examination revealed infiltration with a CD19+, CD20+, and CD5+, but weakly positive CD23 B-cell clone, classified as lymphoplasmacytic lymphoma with typical histology. Further investigation revealed that the B-cell clone carried an MYD88-L265P mutation. The final diagnosis was lymphoplasmacytic lymphoma with cold agglutinins.48 According to treatment recommendations, the patient received 4 doses of 375 mg/m2 rituximab in 4 consecutive weeks and the hemoglobin returned to 13.5 g/dL 6 weeks after infusion was initiated. Reticulocyte counts and haptoglobin level normalized, and, at this writing, the patient had remained in complete hematologic remission for 5 years. The DAT result was still positive for anti-C3d, which is often seen, but in most definitions, is not part of the response criteria for AIHA.21
ITP
ITP is a frequent autoimmune complication of CLL and other non-Hodgkin lymphomas.8,16,19,20,68 The diagnosis should be confirmed by a bone marrow biopsy, according to diagnostic criteria for ITP.6,19,69 This confirmation is important in CLL, because the ITP may mimic bone marrow insufficiency. A discordance between hemoglobin and platelet counts should trigger the biopsy, even if not required by CLL guidelines, as well as further investigations, including serum autoantibody assessment, which is not generally recommended.70 Treatment consists of immunosuppression with steroids, according to ITP guidelines (Figure 1D).69 However, in the case of indolent B-cell lymphomas, the addition of rituximab or combinations thereof may be beneficial because of the expected combined effect on AICs and underlying disease. Given that many patients with lymphoma have immunodeficiencies, intravenous immunoglobulin (IVIG) may also be beneficial. However, IVIG should be reserved for patients in whom the disease is refractory to steroids and/or for bleeding in which a rapid increase in platelets is necessary. In addition, patients with recurrent infections are good candidates for IVIG. Response rates in CLL are ∼50%.20 In nonresponders, treatment with a thrombopoietin agonist should be considered. Response rates with eltrombopag in secondary ITP are as high as 81%.69-71 Rituximab with cyclophosphamide and dexamethasone is a good option for targeting both the AIC and the lymphoma.30,31
Patient 4
A 78-year-old man was diagnosed with CLL in September 2012. At this time, he had a WBC of 41 × 109/L. The patient was in good condition with a hemoglobin level of 12.3 g/dL. However, a platelet count of 38 × 109/L showing thrombocytopenia was noted, without symptoms of bleeding. The discrepancy between the almost normal hemoglobin and the low platelet count triggered ITP diagnostics. No platelet-specific antibodies were found, but reticulated thrombocytes were clearly elevated (30.24%; normal values, 2% to 9%). Thrombopoietin levels were within normal range (285 pg/mL; normal, >150 pg/mL). These results, although not diagnostic, were compatible with increased platelet destruction. No anti-platelet antibodies were detected in the peripheral blood. A bone marrow biopsy specimen indicated nodular infiltration with CLL cells (20%), but normal-to-elevated thrombopoiesis compatible with CLL and ITP. According to the International Workshop on Chronic Lymphocytic Leukemia Guidelines the patient was not treated for CLL in the absence of symptoms.18 The patient received 1 mg/kg of prednisolone for 3 days in preparation for surgery, and the platelet count increased to 101 × 109/L. Two years later, the patient had an episode of severe gastrointestinal bleeding. The platelet count was 20 × 109/L with a WBC of 66 × 109/L. A bone marrow biopsy was not performed at this time because of the patient’s preference. Therefore, treatment with 6 cycles of rituximab+cyclophosphamide and dexamethasone (rituximab 375 mg/m2; cyclophosphamide, 750 mg/m2; dexamethasone, 12 mg absolute; 5 days) was chosen as the first-line therapy, with the rationale that it would have a substantial effect on both conditions.29,30 As a result, his blood count returned to near normal values with WBC count, 7 × 109/L; hemoglobin level, 12.4 g/dL; and platelets, 107 × 109/L.
This case shows the efficacy of both steroid monotherapy and rituximab combination therapy (against both CLL and ITP). Although not all ITP guidelines recommend a bone marrow biopsy for diagnosis, it seems important in patients with an underlying lymphoma condition to determine the predominant reason for the thrombocytopenia.
Evans syndrome
Evans syndrome is typically diagnosed in the fifth to sixth decades and is secondary to underlying disorders, including lymphoproliferative diseases in 27% to 50% of cases.72 It has been reported in up to 2.9% of a cohort of patients with CLL.73
Treatment is similar to that of secondary AIHA or ITP and includes the whole spectrum of immunosuppression and antilymphoma therapy.7
Autoimmune granulocytopenia
Secondary autoimmune granulocytopenia or neutropenia (AIG) in lymphoma is rare. AIG should be suspected in patients who have a declining neutrophil count without other causes (eg, myelodysplastic syndromes, drugs, accompanying rheumatic diseases, or infections).65,74 Typical examples include neutropenia in CLL, in T-cell large granulocytic leukemia, or after CAR T-cell therapy. Persistence of isolated neutropenia after chemotherapy or immunotherapy for longer than expected should trigger a diagnostic workup. AIG is sometimes hard to differentiate from long-term toxicities, such as in CLL treated with fludarabine, cyclophosphamide, and rituximab.20 In particular, late-onset neutropenia after anti-CD20 antibody therapy should be considered. In this case, bone marrow investigation and flow cytometry show a reduced number of B-cells, T-lymphocyte imbalances, and lower proportions of myeloid progenitor cells.75 Clinical diagnosis by exclusion and testing for antineutrophil antibodies is usually negative. Neutropenia may persist without long-lasting symptoms, but infections are imminent. In 1 study, 4% of patients with CLL-associated AIG presented with infections.10 Treatment options include granulocyte colony-stimulating factor and immunosuppressive therapy (steroids).76 In T-cell large granulocytic leukemia, the neutropenia frequently responds to low-dose methotrexate, cyclophosphamide, or cyclosporin A.74,77 Rituximab, when used in the setting of rheumatic diseases, is also effective.78
Autoimmune complications of antilymphoma treatments
AICs may occur after treatment with agents other than chemoimmunotherapy or BTK or BCL2 inhibitors, as described earlier.79
Stimulation of the immune system may have detrimental effects on a preexisting AIC. Patients with autoimmune diseases were excluded from most studies of immunomodulators or immune checkpoint inhibitors. We therefore have little knowledge about the actual effects of those agents. Nevertheless, we are very cautious with immunomodulators, such as lenalidomide or phosphatidylinositol 3-kinase inhibitors in patients with preexisting AICs, and, in most cases, checkpoint inhibitors are avoided if possible. At the least, close monitoring is advised. Immune checkpoint inhibitors have hematologic and nonhematologic autoimmune side effects.80-82
Finally, novel cellular therapies such as CAR T-cell treatment targeting CD19 can cause prolonged cytopenias that are not attributable to late effects of chemotherapy or lymphodepletion. This effect should be differentiated from late myelodysplastic syndrome.83,84
Other autoimmune complications
Hematologic AICs sometimes manifest themselves as coagulopathies. This may present with a bleeding tendency, as is the case in factor X deficiency, von Willebrand’s disease in lymphoplasmacytic lymphoma, or acquired hemophilia.85,86 We have also noted a close association of lupus anticoagulants with splenic marginal zone lymphomas that lead to frequent thrombotic events requiring prophylaxis or treatment.5
Nonhematologic AICs may precede the diagnosis of lymphoma for many years. These include Hashimoto’s thyroiditis, rheumatologic diseases such as systemic lupus erythematosus or rheumatoid arthritis, and neurologic diseases, such as polyneuropathy or gastrointestinal problems (celiac disease).6 C1-esterase deficiency is sometimes associated with lymphoma.87 Most of these diseases are B-cell driven and may also be asymptomatic. In many cases, workup of the autoimmune disease will include the search for malignancies, lymphomas in particular.
Recently, the SARS-CoV2 pandemic and the development of efficient vaccines has posed new questions regarding the induction or exacerbation of AICs. Currently, vaccination is recommended for patients who have AICs, but with special attention to those patients. We ask our patients to have their blood counts checked within 1 week after the vaccination whenever possible.88
Course of AICs during and after antilymphoma treatment
The response of the AIC to antilymphoma treatment is dependent on the underlying AIC and the type of treatment. In many instances, the autoantibodies and/or the clinical symptoms respond to antilymphoma treatment. Steroids and CD20 antibodies such as rituximab or obinutuzumab are effective against many autoimmune diseases.
In a series of patients with lymphoma who received R-CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone+rituximab) we documented the disappearance of rheumatic symptoms and rheumatoid arthritis antibodies.89 Similar results were obtained for acquired C1-esterase inhibitor deficiency.87 As discussed, ibrutinib and venetoclax seem to be beneficial, in part by targeting both the lymphoma and the AIC.34-4190
Conclusions and future developments
AICs can occur at any time before, during, or after lymphoma diagnosis and treatment. They may be the indication for treatment by either immunosuppression or antineoplastic agents. Treatment is tailored toward the predominant disease (autoimmune or neoplastic) or both, usually according to their respective guidelines or recommendations. Anti-CD20 antibodies have beneficial effects in B-cell lymphomas. Novel agents, such as B-cell receptor inhibitors, offer new possibilities for treatment. Some of these agents are even tested in specific trials against AICs. However, AICs may also be triggered by some new treatment approaches against lymphoma and will require adaptive clinical management.
Acknowledgments
The authors thank Michaela Bronhagl for expert assistance, and Cathrin Skrabs and Christian Sillaber for advice.
This work was supported by the Vienna Science and Technology Fund (WWTF) Precision Medicine Program LS16-034 (U.J.).
Authorship
Contribution: U.J. and E.P. designed the manuscript, provided the cases, and wrote the manuscript.
Conflict-of-interest disclosure: U.J. has received honoraria and research funding from AbbVie, Bristol Myers Squibb/Celgene, Gilead, Janssen, Novartis, Roche, Sandoz, and Sanofi. E.P. declares no competing financial interests.
Correspondence: Ulrich Jäger, Division of Hematology and Hemostaseology, Department of Medicine I, Medical University of Vienna, Waehringer Guertel 18-20, A-1090 Vienna, Austria; e-mail: ulrich.jaeger@meduniwien.ac.at.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal