

In this issue of Blood, Ji et al1 identified that up-front dual therapy sirolimus plus prednisolone induced faster resolution of life-threatening Kasabach-Merritt phenomenon (KMP) in children with kaposiform hemangioendothelioma (KHE) compared with sirolimus monotherapy.
The investigators successfully completed a large prospective randomized trial on this rare vascular tumor that has captivated hematologists for its vicious infiltrative response to inflammation and its catastrophic intratumoral coagulopathy2,3 (see figure panel A). Their work highlights an impressive collaboration to standardize and coordinate care for a rare disease among 5 institutions throughout western China. It may help us to better understand the unique relationship between inflammation and KHE tumor pathophysiology, ultimately serving to guide optimal medical therapy.
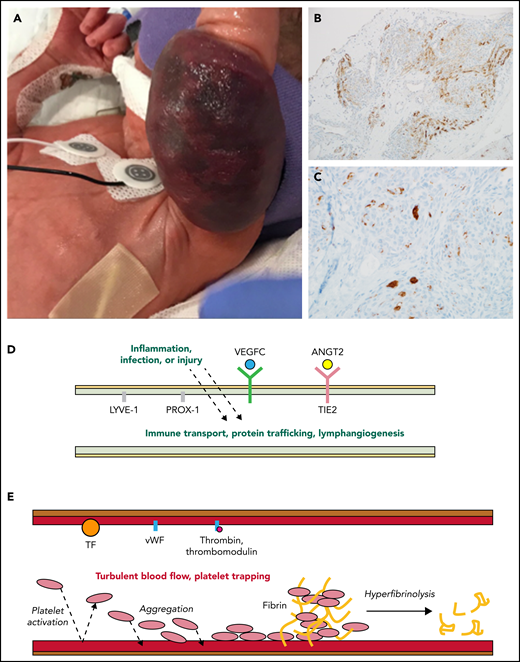
Activated lymphatic and capillary vessels in KHE with KMP. (A) Rapid expansion and infiltration of KHE in response to inflammation, infection, or injury. (B) D2-40+ histologic stain ×20 view reveals abnormal lymphatic vessels. (C) CD61+ histologic stain ×40 view reveals platelet trapping within abnormal capillary vessels. (D) Upregulated and overexpressed signaling from activated abnormal lymphatic endothelium results in increased vessel immune transport, protein trafficking, and lymphangiogenesis. (E) Turbulent blood flow within abnormal capillary vessels results in activated endothelium, consumptive platelet trapping, and hyperfibrinolysis causing KMP. ANGT2, angiopoietin-2; TF, tissue factor; vWF, von Willebrand factor.
Activated lymphatic and capillary vessels in KHE with KMP. (A) Rapid expansion and infiltration of KHE in response to inflammation, infection, or injury. (B) D2-40+ histologic stain ×20 view reveals abnormal lymphatic vessels. (C) CD61+ histologic stain ×40 view reveals platelet trapping within abnormal capillary vessels. (D) Upregulated and overexpressed signaling from activated abnormal lymphatic endothelium results in increased vessel immune transport, protein trafficking, and lymphangiogenesis. (E) Turbulent blood flow within abnormal capillary vessels results in activated endothelium, consumptive platelet trapping, and hyperfibrinolysis causing KMP. ANGT2, angiopoietin-2; TF, tissue factor; vWF, von Willebrand factor.
KHEs are histologically composed of blood vessels and lymphatic channels (characteristically staining D2-40+ or lymphatic vessel endothelial receptor-1 (LYVE-1, and CD34+), and disrupted angiogenesis and lymphangiogenesis are key to the tumor’s pathophysiology2,3 (see figure panels B and D). The lymphatic system is exquisitely sensitive to local and systemic inflammatory triggers, making it interesting to hypothesize how these pathways adapt when lymphatic channels are incorporated throughout a vascular tumor. The vascular endothelial growth factor C (VEGFC)/VEGFR3 and angiopoietin-2/TIE2 endothelial specific receptor tyrosine kinase (TIE2) signaling pathways within lymphatic endothelial cells are critical for regulating angiogenesis, lymphangiogenesis, and vascular stability.3,4 These endothelial cell signaling pathways are likely to be key in promoting tumor inflammation, invasiveness, and growth.3 Supporting the evidence for the role of these pathways in KHE pathogenesis are studies showing that prospero homeobox protein 1 (Prox1) overexpression can induce an invasive vascular tumor phenotype in mice, there is increased VEGFR3 expression in KHEs compared with other benign vascular tumors , and high serum levels of angiopoietin-2 are seen in patients with KHE and a decrease in response to therapy.3
In response to inflammation, trauma, or infection, KHEs uniquely erupt to aggressively infiltrate into surrounding tissues and entrap the elements of primary hemostasis in a cycle of consumptive hyperfibrinolysis called KMP. Clinically, KMP presents itself as a profound and sustained thrombocytopenia and a consumptive coagulopathy associated with a significant bleeding risk and a high risk for mortality.2 Platelet trapping within the tumor is known to occur in tumors, both with and without KMP3 (see figure panel C). Podoplanin expressed on lymphatic endothelial cells may interact with C-type lectin receptors on platelets, contributing to platelet aggregation in the tumor.3 Endothelial cell damage within abnormal vessels of the tumor, via the exposure of the extracellular matrix, release von Willebrand factor and activated tissue factor.5 Together, inflammation-induced release of these factors promotes rapid platelet and fibrinogen consumption and an increase in fibrin-split products (D-dimer), tumor expansion, and clinical bleeding (see figure panel E).5 Intratumoral hemorrhage is certainly part of tumor expansion in patients with severe KMP, but there is also evidence from the oncology literature that platelet-released factors contribute to angiogenesis within the tumor.6 The primary objective when initiating therapy is rapid resolution of life-threatening KMP and clinical bleeding.
Ji et al take on the problem that determination of optimal first-line therapy has been limited by the lack of prospective randomized trials for this rare tumor. Initial consensus-derived standards of practice were adapted from historic oncologic practices and consisted of dual-therapy vincristine and steroids as first-line therapy for KHE with KMP.7 However, the first prospective trial on the use of sirolimus in patients with vascular anomalies, including KHE, was published 3 years later and generated much excitement about the efficacy of mTOR inhibition in KHE.8 Since that time, the use of sirolimus in KHE has been reported in several case series and retrospective studies. Faced with life-threatening hemorrhage, many use a combination of medical therapies, including sirolimus, steroids, vincristine, and antiplatelet agents. The impact of steroids continues to be debated, because they are often thought of as the best and worst immunosuppressive agents in our armamentarium. Wide availability and rapid onset of action make steroids a reliable up-front agent. However, the requirement for steroids as part of therapy for longer-term outcomes has not been known. In this study, Ji et al demonstrate that up-front steroid therapy improves time to resolution of KMP, as well as durability of platelet response. Overall lesion response was also improved in the group receiving up-front steroids with sirolimus. This effect was seen as far out as 12 months following the initiation of therapy. Importantly, this benefit was seen without an increase in infectious complications, which is 1 of the arguments against steroid use in infants and young children.
Ji et al speculate that the combined neoplastic and inflammatory nature of KHE is best treated with pharmacotherapy that targets both key pathologic features of this vascular tumor. Indeed, there are data from the literature on infantile hemangiomas that corticosteroids suppress VEGF signaling and may suppress other proangiogenic factors.9 Corticosteroids have also been found to have inhibitory effect on angiopoietin-2 expression in endothelial cells and in other tumor types.10 It is possible that early blunting of lymphangiogenesis within the tumor prevents rapid tumor expansion and the cycle of inflammation, activation of coagulation, and tumor growth, thus improving near-term and longer-term outcomes.
Inflammation, disrupted vasculogenesis, and severe consumptive coagulopathy are all key to the dangerous pathophysiology and aggressive presentation of KHE. Prospective treatment and risk-stratification studies, such as performed by Ji et al, are needed to better understand this rare tumor and improve patient outcomes, but the lessons learned from such endeavors are not unique to KHE. Identification of the complex mechanisms regulating angiogenesis, lymphangiogenesis, and disrupted coagulation and inflammation at the endothelial cell level are key to improving our understanding of many disease processes and malignancies. As novel therapeutic agents are identified that target these pathways, treatment options for patient with rare tumors like KHE will expand.
Conflict-of-interest disclosure: The authors declare no competing financial interest.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal