

Key Points
ACKR3/CXCR7 ligation regulates COX-1 and 12-LOX–derived prothrombotic lipids but favors antiplatelet lipids that trigger platelet inhibition.
Contrary to prothrombotic CXCR4, ACKR3/CXCR7 modulates thrombotic and thromboinflammatory functions without affecting hemostasis.

Abstract
Platelet ACKR3/CXCR7 surface expression is enhanced and influences prognosis in coronary artery disease (CAD) patients, who exhibit a distinct atherothrombotic platelet lipidome. Current investigation validates the potential of ACKR3/CXCR7 in regulating thromboinflammatory response through its impact on the platelet lipidome. CAD patients with enhanced platelet ACKR3/CXCR7 expression exhibited reduced aggregation. Pharmacological CXCR7 agonist (VUF11207) significantly reduced prothrombotic platelet response in blood from acute coronary syndrome patients ex vivo. CXCR7 agonist administration reduced thrombotic functions and thromboinflammatory plateletleukocyte interactions post–myocardial infarction and arterial injury in vivo. ACKR3/CXCR7 ligation did not affect surface availability of surface receptors, coagulation profile, bleeding time, plasma-dependent thrombin generation (thrombinoscopy), or clot formation (thromboelastography) but counteracted activation-induced phosphatidylserine exposure and procoagulant platelet-assisted thrombin generation. Targeted (micro-UHPLC-ESI-QTrap-MS/MS) and untargeted (UHPLCESI-QTOF-MS/MS) lipidomics analysis revealed that ACKR3/CXCR7 ligation favored generation of antithrombotic lipids (dihomo-γ-linolenic acid [DGLA], 12-hydroxyeicosatrienoic acid [12-HETrE]) over cyclooxygenase-1 (COX-1) or 12-lipoxygenase (12-LOX) metabolized prothrombotic and phospholipase-derived atherogenic lipids in healthy subjects and CAD patients, contrary to antiplatelet therapy. Through 12-HETrE, ACKR3/CXCR7 ligation coordinated with Gαs-coupled prostacyclin receptor to trigger cyclic adenosine monophosphate/protein kinase A–mediated platelet inhibition. ACKR3/CXCR7 ligation reduced generation of lipid agonists and lipid signaling intermediates, which affected calcium mobilization, intracellular signaling, and consequently platelet interaction with physiological matrices and thromboinflammatory secretome. This emphasized its functional dichotomy from prothrombotic CXCR4. Moreover, CXCR7 agonist regulated heparin-induced thrombocytopenia–sera/immunoglobulin G–triggered platelet and neutrophil activation, heparin-induced platelet aggregation, generation of thromboinflammatory lipids, platelet-neutrophil aggregate formation, and thromboinflammatory secretion ex vivo. Therefore, ACKR3/CXCR7 may offer a novel therapeutic strategy in acute/chronic thromboinflammation exaggerated cardiovascular pathologies and CAD.
Introduction
Thromboinflammatory attributes of platelets are widely investigated in cardiovascular disease to find novel therapeutic interventions.1,2 Immunothrombosis resulting from FcγRIIA-mediated3 platelet activation and involving thromboinflammatory platelet-leukocytes associations4 may manifest venous, arterial, and pulmonary microvascular thrombosis,5,6 as well as thromboischemic complications.5 Platelet-monocyte interactions in acute coronary syndrome (ACS)7 and platelet-neutrophil associations in heparin induced thrombocytopenia (HIT)8 aggravate disease severity and thrombotic disposition. Platelet-derived thromboinflammatory mediators (eg, IL-1β,sCD40L) add to circulatory levels during acute inflammation6,9 and chronic atheroprogression.10 Given the unmet need for better antiplatelet strategies in thromboischemic pathologies to overcome the drawbacks of current therapeutics10 and inefficacy of aspirin (ASA)11 in immunothrombosis-associated mortality, innovative antithromboinflammatory approaches1,5,10 are warranted.
Thromboinflammatory platelet functions propagate vascular and microvascular thrombosis in veins,8 arteries,12 and pulmonary micro-capillaries,13 as well as influence the severity of target organ damage also subsequent regeneration, remodeling, and functional recovery.1,14 Coronary artery disease (CAD) patients show increased surface expression of the chemokine stromal cell–derived factor CXCL12/SDF1α15 and its receptors, CXCR4 and ACKR3/CXCR7, on platelets.16 ACKR3/CXCR7 surface expression is particularly enhanced in ACS patients and associated with improved left ventricular ejection fraction (LVEF) and prognosis.16,17 Interestingly, ACKR3/CXCR7 is richly expressed by cardiomyocytes; it is significantly enhanced at the infarct border zone of the affected myocardium in mice18 and in heart failure patients.18 Endothelial deletion of ACKR3/CXCR7 increases infarct size and mortality and aggravates functional impairment post-MI,19 whereas targeted delivery of cxcr7 gene19 and administration of CXCR7 agonist AMD310020 (CXCR4 antagonist) or TC1401218,19,21 (CXCR4 inverse agonist) offers cardio-protective benefits19,21 in mice. However, the implications of enhanced platelet ACKR3/CXCR7 availability among ACS patients16,17 in modulating thrombotic and thromboinflammatory actions post–myocardial infarction/reperfusion injury (MI/RI) remained unexplored. This is of significance because activated platelets infiltrate the myocardium with either deleterious or regenerative consequences, as well as cause recurrent atherothrombotic and thromboischemic complications.22
Plasma levels of physiological ligands CXCL12/SDF1α23,24 and macrophage migration inhibitory factor (MIF)25 are progressively elevated with disease severity in ACS patients, which may influence platelet CXCR4 and ACKR3/CXCR7 availability26,27 and thereby their pathophysiological engagement post-MI. CXCL12/SDF1α and MIF promote platelet survival through ACKR3/CXCR7.26,27 Plasma-CXCL12/SDF1α may exert a prothrombotic effect through platelet CXCR4, but MIF does not regulate platelet response to external stimuli.27,28 However, it reduces externalization of thrombogenic phospholipid phosphatidylserine on procoagulant platelets, which consequently exercises an antithrombotic effect, counteracted upon blocking ACKR3/CXCR7.27 On the contrary, prothrombotic CXCL12/SDF1α29 promotes collagen-induced platelet aggregation, adenosine triphosphate (ATP) release, thromboxane production and thrombus formation30 through intracellular calcium mobilization, triggering activatory signaling cascade involving phosphoinositide 3-kinase (PI3K), Akt, PDK1, glycogen synthase kinase 3 beta, and myosin light chain phosphorylation.31 These studies suggest the functional dichotomy of CXCR4 and ACKR3/CXCR7, the balance of which may direct the course of thromboinflammation and thrombotic propensity in cardiovascular pathologies arising from chronic10 and acute inflammation.5
Previous investigators have employed cyclic peptide (TC14012), allosteric agonist (AMD3100), and agonist (CCX771)32 to demonstrate the therapeutic implications of ACKR3/CXCR7 in myocardial infarction,19,21 pulmonary fibrosis,33 and atherosclerosis.34 We have used the high-affinity (pKi = 8.1)32,35-37 CXCR7 agonist (VUF11207) instead of proinflammatory physiological ligands to validate the potential of ACKR3/CXCR7 in governing thrombotic and thromboinflammatory platelet functions post-MI, following arterial injury, and those induced by HIT-sera/immunoglobulin G (IgG) acting through FcγRIIA. In search of its mechanism of action we also revealed its regulatory impact on the platelet lipidome.
Methods
A detailed description is provided in supplemental Methods; experimental conditions are specified in figure legends.
Platelet functions
Degranulation (CD62P, CD63 surface expression by flow cytometry, ATP release by lumi-aggregometry38), αIIbβ3-integrin activation (flow cytometry),1,8 spreading, elastic modulus39 (scanning ion conductance microscopy [SICM]), aggregation (impedance29 and lumi-aggregometry38), procoagulant platelets (flow cytometry),27 thrombus formation1,40 (total thrombus analysis system [T-TAS]), intraplatelet calcium mobilization (flow cytometry),41 platelet-leukocyte aggregates (flow cytometry), and thromboinflammatory platelet release (cytometric bead array) were evaluated in presence/absence of CXCR7 agonist/vehicle control.
Hemostatic and coagulation parameters
Lipidomics
Untargeted (UHPLC-ESI-QTOF-MS/MS) and targeted (micro-UHPLC-ESI-QTrap-MS/MS) lipidomics analyses for oxylipins43,44 were performed for resting and thrombin-activated platelets45 and platelet supernatant/releasates46 in presence/absence of CXCR7 agonist/vehicle control. Lipid extraction from platelet releasate was done with MTBE/MeOH/H2O; from platelet pellet in a monophasic extraction with isopropanol/water 9:1 (vol/vol), followed by solid-phase extraction on Bond Elut Certify II cartridges (Agilent) with ethyl acetate/n-hexane/acetic acid 75:24:1 (vol/vol/v) for oxylipins. 1290-Agilent UHPLC instrument, PAL-HTX xt DLW autosampler (CTC Analytics) and SCIEX TripleTOF 5600+ were used for untargeted lipidomics; Eksigent MicroLC 200 Plus System (Sciex) and QTrap 4500 MS instrument (Sciex) were used for targeted (micro-UHPLC-ESI-QTrap-MS/MS) lipidomics analysis. MS-Dial47 was used for peak picking, lipid identification supported with confirmation of correct retention time,45 manual curation, and peak integration.
cAMP levels
Time-dependent increase in intraplatelet cyclic adenosine monophosphate (cAMP) levels following CXCR7 agonist treatment was ascertained by LC-ESI-MS/MS.
Immunoblots
Western blots for phosphorylated phospholipase C gamma (PLCγ), Src Family Tyr416, protein kinase C (PKC), PI3K, Akt, and p38MAPK were performed in collagen-related peptide (CRP)-activated platelets in presence/absence of CXCR7 agonist/vehicle control.26,27
CAD patient cohort
Blood was collected from femoral arterial access site of patients during coronary angiography16,48 (Tables 1-3). All patients admitted to the Department of Cardiology and Angiology gave written informed consent. The study was approved by the institutional ethics committee (270/2011BO1, 237/2018BO2) and complied with the Declaration of Helsinki and Good Clinical Practice guidelines.
Baseline characteristics of CAD patients enrolled for platelet aggregation test and surface expression of CXCR4 and ACKR3/CXCR7
| CAD patients’ characteristics | CAD (n = 230) |
|---|---|
| Age (mean ± SD) | 67.1 (± 11.9) |
| Male gender | 170 (73.9%) |
| LVEF % at admission (mean ± SD) | 50.6 (± 11.0) |
| Cardiovascular risk factors | |
| Arterial hypertension | 200 (87.0%) |
| Hyperlipidemia | 134 (58.3%) |
| Diabetes mellitus type 2 | 60 (26.1%) |
| Smoking | 104 (45.2%) |
| Medication on admission | |
| ASA | 123 (53.5%) |
| Clopidogrel | 27 (11.7%) |
| Prasugrel | 4 (1.7%) |
| Ticagrelor | 10 (4.3%) |
| ACE inhibitors | 90 (39.1%) |
| ARBs | 38 (16.5%) |
| β blockers | 117 (50.9%) |
| Ca-channel inhibitors | 45 (19.6%) |
| Diuretics | 74 (32.1%) |
| Statins | 99 (43.0%) |
| Reason of admission/clinical diagnosis | |
| ACS | 142 (61.7%) |
| CCS | 88 (38.3%) |
| Plasma lipid profile | |
| Total cholesterol (mg/dl) | 177.1 (± 46.5) |
| CAD patients’ characteristics | CAD (n = 230) |
|---|---|
| Age (mean ± SD) | 67.1 (± 11.9) |
| Male gender | 170 (73.9%) |
| LVEF % at admission (mean ± SD) | 50.6 (± 11.0) |
| Cardiovascular risk factors | |
| Arterial hypertension | 200 (87.0%) |
| Hyperlipidemia | 134 (58.3%) |
| Diabetes mellitus type 2 | 60 (26.1%) |
| Smoking | 104 (45.2%) |
| Medication on admission | |
| ASA | 123 (53.5%) |
| Clopidogrel | 27 (11.7%) |
| Prasugrel | 4 (1.7%) |
| Ticagrelor | 10 (4.3%) |
| ACE inhibitors | 90 (39.1%) |
| ARBs | 38 (16.5%) |
| β blockers | 117 (50.9%) |
| Ca-channel inhibitors | 45 (19.6%) |
| Diuretics | 74 (32.1%) |
| Statins | 99 (43.0%) |
| Reason of admission/clinical diagnosis | |
| ACS | 142 (61.7%) |
| CCS | 88 (38.3%) |
| Plasma lipid profile | |
| Total cholesterol (mg/dl) | 177.1 (± 46.5) |
We included n = 230 consecutive patients with symptomatic CAD to analyze platelet ACKR3/CXCR7 surface expression and aggregation response to ADP and TRAP in whole blood samples.
HIT-associated thromboinflammation
Sera and corresponding IgG fractions from 3 patients with clinically and serologically confirmed HIT-associated thrombosis (using 4Ts score 4-6, enzyme-linked immunosorbent assay (ELISA), heparin-induced platelet aggregation [HIPA]-positive) were collected to evaluate platelet and neutrophil activation (flow cytometry), platelet-neutrophil aggregates (flow cytometry) thromboxane A2 (TxA2), 12-HETE generation (lipidomics), and platelet thromboinflammatory release (cytometric bead array).
Animal experimentations
Eight- to 10-week-old C57BL/6J mice of either sex (Jackson Laboratories) were used for ex vivo platelet function assays, tail-bleeding time, coagulation profile (PT, APTT), FeCl3-induced arterial thrombosis,27,29 and MI/RI model by left anterior descending artery ligation.49 All experimentations were conducted according to the German law for welfare of animals, following Animal Research: Reporting of In Vivo Experiments guidelines, and approved by local authorities.
Statistical analysis
Experimental studies
Data (mean ± standard error of the mean [SEM]/standard deviation [SD]) were analyzed using GraphPad Prism software (GraphPad Software, Inc.; San Diego, CA, USA) at P < 0.05 statistical significance with analysis of variance (ANOVA) for >2 groups; Mann-Whitney U test, Welch’s t test, and Wilcoxon matched-pairs signed rank test for 2 groups.
Clinical cohort
Data (median ± SD) were analyzed using SPSS version 21.0 (SPSS, Inc.; Chicago, IL, USA). Nonnormally distributed data were compared using the Mann-Whitney U test.
Results
ACKR3/CXCR7 may exert antithrombotic actions post-MI/RI
Increased platelet ACKR3/CXCR7 surface expression in ACS patients is associated with improved prognosis.16,17 Symptomatic CAD patients (n = 230; Table 1) with relatively enhanced platelet ACKR3/CXCR7 surface expression showed significantly lower aggregation response to adenosine 5′-diphosphate (ADP) and TRAP (Figure 1A), which paved the way to assess the antithrombotic influence of platelet ACKR3/CXCR7. Pharmacological CXCR7 agonist (VUF11207) induced a dose- and time-dependent internalization of ACKR3/CXCR7 (Figure 1Bi-Bii) in vitro, as expected,35 but contrary to CXCL12/SDF-1α,26,27 which externalizes ACKR3/CXCR7. However, unlike CXCL12/SDF-1α,26,27 CXCR7 agonist (VUF11207) neither internalized CXCR4 (supplemental Figure 1Ai-ii) nor affected the surface expression of other CXCR-GPCRs (ie, CXCR1, CXCR2, CXCR6) on human platelets vs vehicle control (supplemental Figure 1Bi-iii), suggesting an ACKR3/CXCR7-specific effect. Similarly, a time-dependent internalization of murine platelet ACKR3/CXCR7 was observed in vitro (supplemental Figure 1C) and among circulatory platelets following CXCR7 agonist administration (until 3 hours) in vivo (supplemental Figure 1D), without affecting CXCR4 surface expression (supplemental Figure 1C-D) vs vehicle control.
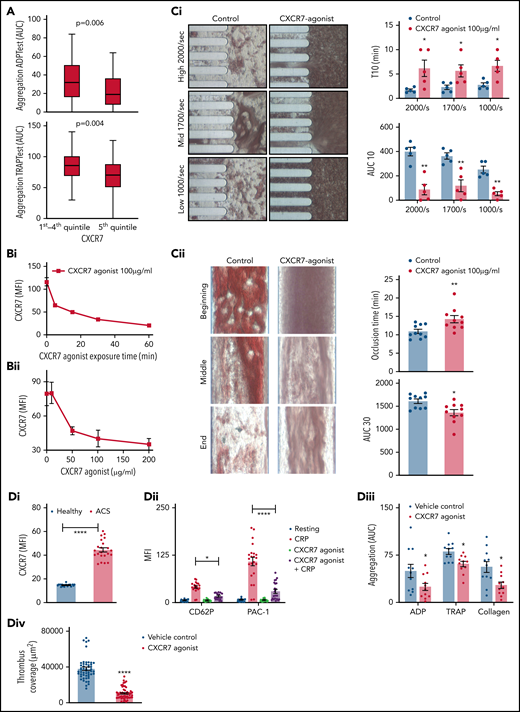
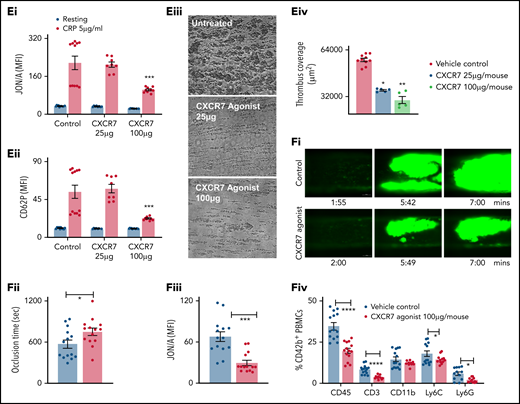
ACKR3/CXCR7 exerts antithrombotic effects. (A) Aggregation response to ADP (ADPTest, Roche) and TRAP-6 (TRAPTest, Roche) evaluated by impedance aggregometry in whole blood samples acquired in hirudinized tubes (Sarstedt) from CAD (n = 230; ACS, n = 142; CCS, n = 88) patients. P values using Mann-Whitney U test (line in box plots denote median). Flow cytometric detection of ACKR3/CXCR7 (mouse anti-human/mouse ACKR3/CXCR7-PE) internalization from the platelet surface following CXCR7 agonist treatment in a (Bi) time and (Bii) dose-dependent manner, gating for platelet-specific marker CD42b (anti-human CD42b-FITC) in whole blood. Data in the graphs are mean ± SEM from 4 healthy donors. T-TAS (Fujimori Kogyo Co Ltd, Shinjuku, Japan) data showing (Ci) thrombus coverage (AUC) and time to attainment of 10 kPa (T10) pressure over collagen (PL-chip) using 320 µL of hirudin anticoagulated blood and (Cii) thrombus coverage (AUC) and time to occlusion over collagen + tissue factor (AR-chip) using 450 µL of recalcified citrated human blood at arterial shear rates in presence/absence of CXCR7 agonist (100 µg/mL) or vehicle control incubated for 30 minutes at room temperature before perfusion. Data in the graphs are mean ± SEM from 5 experiments with healthy donors. *P < .05, **P < .01 using Mann-Whitney test. (Di) Surface expression of ACKR3/CXCR7 on platelets in n = 11 ACS patients as compared with healthy subjects (n = 11). Data in the graphs are mean ± SEM. **** P < .0001 using Mann-Whitney U test. Ex vivo whole blood functional assays performed with blood collected from ACS patients (n = 11) showing (Dii) surface expression of CD62P (anti-human CD62P-FITC) and PAC-1 (PAC-1–FITC) binding detected by flow cytometry gating for CD42b+-platelet (anti-human CD42b-PE) population; (Diii) aggregation response to ADP (ADPTest), TRAP-6(TRAPTest), and collagen(ColTest) in hirudinized blood; and (Div) thrombus formation over collagen coated (100 µg/mL) surface in parallel plate flow chamber assay in presence/absence of CXCR7 agonist (100 µg/mL)/vehicle control preincubated for 30 minutes at room temperature. *P < .05, **** P < .0001 using Mann-Whitney U test between 2 groups and ANOVA followed by Sidak’s multiple comparison test for >2 groups. Ex vivo analysis of murine platelet functions performed 1 hour after administration of CXCR7 agonist (100 µg/mL)/vehicle control (1% DMSO) showing (Ei) JON/A (JON/A-PE) response, (Eii) CD62P (anti-mouse CD62P-FITC) surface expression by whole blood flow cytometry gating for platelet-specific marker CD42b (anti-mouse CD42b-Dylight-649), and (Eiii-Eiv) thrombus coverage over collagen-coated surface (100 µg/mL) in ex vivo parallel plate flow chamber assay. Data in the graphs are mean ± SEM from 5 mice per group. CXCR7 agonist (100 µg per mouse) or vehicle control (1% DMSO) along with in vivo platelet-labeling antibody (GPIbβ-Dylight 488, 0.1 µg/gm by body weight) was administered IV 1 hour prior to surgical procedures to inflict carotid artery injury. Intravital microscopy (IVM) with NIS-Elements (Nikon) microscope was carried out using a 10x objective following carotid artery injury inflicted by application of a filter paper soaked in 15% FeCl3 for 1 minute. (Fi) Reduced thrombus formation (green fluorescence from platelets in circulation stained in vivo with GPIbβ-Dylight 488) and (Fii) time to vessel occlusion (*P = .034). Following IVM analysis, blood was collected to perform further ex vivo analysis of platelet functions. Flow cytometric detection of (Fiii) JON/A response, (Fiv) platelet (anti-mouse GPIbβ-Dylight 488) aggregate formation with leukocytes (anti-mouse CD45-APC), lymphocytes (anti-mouse CD3-APC), monocytes, and neutrophils (anti-mouse Ly6C-APC, anti-mouse Ly6G-PE, anti-mouse CD11b-APC) following carotid artery injury in CXCR7 agonist/vehicle control–administered mice. In panels Ei through Fiii, *P < .05, **P < .01, ***P < .001 using an unpaired Student t test with Welch’s correction; in panel Fiv, *P < .05, ****P < .0001 using Mann-Whitney U test for each surface marker. Data in the graphs are mean ± SEM from 14 mice per group. AUC, area under the curve; APC, apocyanin; DMSO, dimethyl sulfoxide; FITC, fluorescein isothiocyanate.
ACKR3/CXCR7 exerts antithrombotic effects. (A) Aggregation response to ADP (ADPTest, Roche) and TRAP-6 (TRAPTest, Roche) evaluated by impedance aggregometry in whole blood samples acquired in hirudinized tubes (Sarstedt) from CAD (n = 230; ACS, n = 142; CCS, n = 88) patients. P values using Mann-Whitney U test (line in box plots denote median). Flow cytometric detection of ACKR3/CXCR7 (mouse anti-human/mouse ACKR3/CXCR7-PE) internalization from the platelet surface following CXCR7 agonist treatment in a (Bi) time and (Bii) dose-dependent manner, gating for platelet-specific marker CD42b (anti-human CD42b-FITC) in whole blood. Data in the graphs are mean ± SEM from 4 healthy donors. T-TAS (Fujimori Kogyo Co Ltd, Shinjuku, Japan) data showing (Ci) thrombus coverage (AUC) and time to attainment of 10 kPa (T10) pressure over collagen (PL-chip) using 320 µL of hirudin anticoagulated blood and (Cii) thrombus coverage (AUC) and time to occlusion over collagen + tissue factor (AR-chip) using 450 µL of recalcified citrated human blood at arterial shear rates in presence/absence of CXCR7 agonist (100 µg/mL) or vehicle control incubated for 30 minutes at room temperature before perfusion. Data in the graphs are mean ± SEM from 5 experiments with healthy donors. *P < .05, **P < .01 using Mann-Whitney test. (Di) Surface expression of ACKR3/CXCR7 on platelets in n = 11 ACS patients as compared with healthy subjects (n = 11). Data in the graphs are mean ± SEM. **** P < .0001 using Mann-Whitney U test. Ex vivo whole blood functional assays performed with blood collected from ACS patients (n = 11) showing (Dii) surface expression of CD62P (anti-human CD62P-FITC) and PAC-1 (PAC-1–FITC) binding detected by flow cytometry gating for CD42b+-platelet (anti-human CD42b-PE) population; (Diii) aggregation response to ADP (ADPTest), TRAP-6(TRAPTest), and collagen(ColTest) in hirudinized blood; and (Div) thrombus formation over collagen coated (100 µg/mL) surface in parallel plate flow chamber assay in presence/absence of CXCR7 agonist (100 µg/mL)/vehicle control preincubated for 30 minutes at room temperature. *P < .05, **** P < .0001 using Mann-Whitney U test between 2 groups and ANOVA followed by Sidak’s multiple comparison test for >2 groups. Ex vivo analysis of murine platelet functions performed 1 hour after administration of CXCR7 agonist (100 µg/mL)/vehicle control (1% DMSO) showing (Ei) JON/A (JON/A-PE) response, (Eii) CD62P (anti-mouse CD62P-FITC) surface expression by whole blood flow cytometry gating for platelet-specific marker CD42b (anti-mouse CD42b-Dylight-649), and (Eiii-Eiv) thrombus coverage over collagen-coated surface (100 µg/mL) in ex vivo parallel plate flow chamber assay. Data in the graphs are mean ± SEM from 5 mice per group. CXCR7 agonist (100 µg per mouse) or vehicle control (1% DMSO) along with in vivo platelet-labeling antibody (GPIbβ-Dylight 488, 0.1 µg/gm by body weight) was administered IV 1 hour prior to surgical procedures to inflict carotid artery injury. Intravital microscopy (IVM) with NIS-Elements (Nikon) microscope was carried out using a 10x objective following carotid artery injury inflicted by application of a filter paper soaked in 15% FeCl3 for 1 minute. (Fi) Reduced thrombus formation (green fluorescence from platelets in circulation stained in vivo with GPIbβ-Dylight 488) and (Fii) time to vessel occlusion (*P = .034). Following IVM analysis, blood was collected to perform further ex vivo analysis of platelet functions. Flow cytometric detection of (Fiii) JON/A response, (Fiv) platelet (anti-mouse GPIbβ-Dylight 488) aggregate formation with leukocytes (anti-mouse CD45-APC), lymphocytes (anti-mouse CD3-APC), monocytes, and neutrophils (anti-mouse Ly6C-APC, anti-mouse Ly6G-PE, anti-mouse CD11b-APC) following carotid artery injury in CXCR7 agonist/vehicle control–administered mice. In panels Ei through Fiii, *P < .05, **P < .01, ***P < .001 using an unpaired Student t test with Welch’s correction; in panel Fiv, *P < .05, ****P < .0001 using Mann-Whitney U test for each surface marker. Data in the graphs are mean ± SEM from 14 mice per group. AUC, area under the curve; APC, apocyanin; DMSO, dimethyl sulfoxide; FITC, fluorescein isothiocyanate.
CXCR7 agonist showed a dose-dependent (10-200 µg/mL) inhibitory effect on CRP-induced CD62P surface expression, PAC-1 binding (supplemental Figure 2A), collagen-induced aggregation (supplemental Figure 2B), and thrombus formation (supplemental Figure 2C-D) ex vivo. We further explored thrombus formation over collagen-coated (Figure 1Ci; supplemental Videos 1-6) and collagen + tissue factor (TF)-coated chips (Figure 1Cii; supplemental Videos 7-8) with T-TAS. CXCR7 agonist (100 µg/mL) reduced thrombus formation (Figure 1Ci) and decelerated (time to 10 kPa pressure attainment) thrombus build-up over collagen. Similarly, CXCR7 agonist decreased thrombus coverage and prolonged time to occlusion (Figure 1Cii) over collagen++ TF-coated surface.
Next, we assessed the antithrombotic efficacy of CXCR7 agonist in whole blood platelet function assays among ACS patients (n = 11) ex vivo, who exhibited significantly enhanced platelet ACKR3/CXCR7 surface expression (Figure 1Di). CXCR7 agonist counteracted CRP-induced degranulation (CD62P), αIIbβ3-integrin activation (PAC-1) (Figure 1Dii), ADP-, collagen-, TRAP-induced aggregation (Figure 1Diii), and thrombus formation (Figure 1Div). Therefore, enhanced platelet ACKR3/CXCR7 availability in ACS patients16 post-myocardial infarction (MI) may possibly be used to regulate platelet response.
ACKR3/CXCR7 ligation modulates thrombotic and thromboinflammatory functions
We assessed the antithrombotic potential of platelet ACKR3/CXCR7 in regulating arterial thrombosis and platelet functions post-MI using murine models. Since CXCR7 agonist showed a dose-dependent antithrombotic efficacy with murine blood (supplemental Figure 2E) in vitro, we administered CXCR7 agonist at suboptimal (25 µg per mouse) and optimal (100 µg per mouse) doses in vivo and after 1 hour analyzed platelet functions ex vivo. CXCR7 agonist (100 µg per mouse) administration significantly counteracted subsequent CRP-induced αIIbβ3-integrin activation (JON/A-response) (Figure 1Ei), degranulation (CD62P) (Figure 1Eii), and thrombus formation (Figure 1Eiii-Eiv) ex vivo, as well as reduced arterial thrombus formation, significantly prolonging time to vessel occlusion (Figure 1Fi-ii; supplemental Videos 9-10), and countered αIIbβ3-integrin activation (Figure 1Fiii) in vivo. Platelet-leukocyte interactions in circulation substantiate atherothrombosis14 and mediate acute thromboinflammation,4,13 necessitating adequate antiplatelet strategies. CXCR7 agonist administration reduced circulatory platelet-leukocyte aggregate formation (Figure 1Fiv) following arterial injury.
Antiplatelet therapies are cornerstone in preventing platelet hyper-reactivity causing recurrent thromboischemic complications in ACS patients.50,51 Corroborating previous reports using TC14012 (10 mg/kg)19,21 and AMD3100,20 administration of VUF11207 (300 µg per mouse) at a comparable/similar dose reduced infarct size (supplemental Figure 2F) and showed less deteriorated LVEF in mice 24 hours post-MI (ejection fraction/infarct size: vehicle control 2.35 ± 0.82 vs CXCR7 agonist 3.45 ± 1.01, P = .037; infarct area/area at risk: vehicle control 46.65 ± 6.740 vs CXCR7 agonist 33.21 ± 4.087, P = .0005). Circulatory platelet ACKR3/CXCR7 surface expression was enhanced, whereas CXCR4 expression was reduced (Figure 2Ai) 24 hours post-MI, contrary to increment in both CXCR4-ACKR3/CXCR7 on platelets from CAD patients as compared with healthy subjects and significantly increased expression of ACKR3/CXCR7 in ACS patients as compared with stable CAD.16,48 This suggests interspecies differences in relative platelet surface availability of CXCR4-ACKR3/CXCR7. In addition to its known therapeutic effect on the ischemic heart,18,19 CXCR7 agonist administration reduced αIIbβ3-integrin activation (Figure 2Aii), CD62P exposure, platelet surface expression of prothrombotic, inflammatory CXCL12/SDF-1α (Figure 2Aiii) in vivo, and decreased activation potential/responsiveness of circulatory platelets ascertained by CRP-induced activation ex vivo. CXCR7 agonist–treated mice showed reduced thrombotic potential (Figure 2Aiv), declined plasma levels of proinflammatory mediators (IL1α, IL1β, TNFα, IFNγ, IL-6, MCP-1) derived from different cellular origins (eg, circulatory cells), and not exclusively platelets (Figure 2B), as well as decreased thromboinflammatory platelet-leukocyte aggregates in circulation (Figure 2C). However, platelet counts, mean platelet volume (Figure 2Di-ii), and surface expression of receptors glycoprotein Ibα, GPV, GPVI, GPIX, αv-integrin, and β3-integrin (Figure 2E) remained unaffected. These experimental (in vivo) and clinical data from ACS patients (ex vivo) suggest that enhanced platelet ACKR3/CXCR7 availability post-MI may be used to counter thrombotic and thromboinflammatory functions.
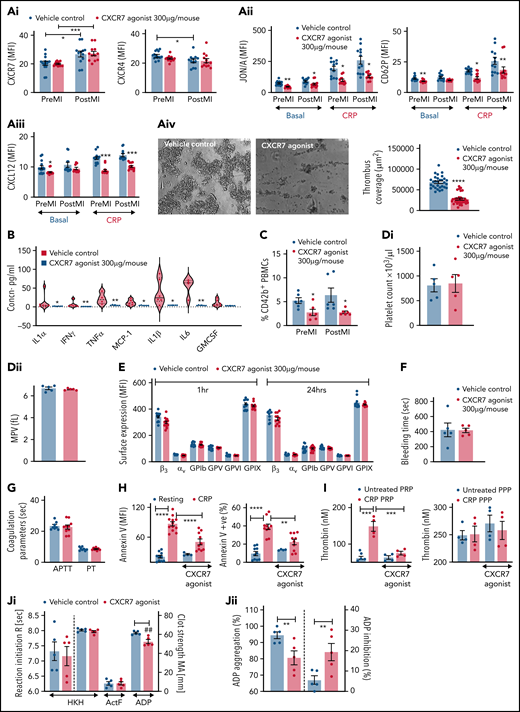
CXCR7 agonist modulates thrombotic and thromboinflammatory platelet response without compromising plasma dependent coagulation. CXCR7 agonist (300 µg/mouse) or vehicle control (2% DMSO) was administered IV 1 hour prior to the surgical procedure. Surface expression of (Ai) ACKR3/CXCR7 (anti-human/mouse CXCR7-PE) and CXCR4 (anti-mouse CXCR4-Fluorescein) on circulating murine platelets. (Aii) Platelet JON/A response (JON/A-PE) and CD62P (anti-mouse CD62P-FITC) surface expression and (Aiii) surface expression of CXCL12/SDF-1α (anti human/mouse CXCL12/SDF-1α- Fluorescein) (denoting degranulation) in basal and CRP-activated (incubated ex vivo for 30 minutes at room temperature) state were evaluated by flow cytometry using blood collected pre-MI (before beginning the surgical procedure) and 24 hours post-MI/RI. (Aiv) Thrombus coverage ex vivo. (B) Levels of proinflammatory mediators in plasma (violin plot with median line) measured with cytometric bead arrays (Legendplex murine 13-plex inflammation panel) 24 hours post-MI/RI. (C) Percentage of CD42b+ (anti-mouse CD42b-FITC)-CD45+ (anti-mouse CD45-APC) platelet-leukocyte aggregates in blood collected pre-MI and 24 hours post-MI/RI. In panels A through C, *P < .05, **P < .01, ***P < .001, ****P < .0001 using Mann-Whitney U test. (Di-Dii) Platelet count and mean platelet volume (MPV) 24 hours post-MI/RI. CXCR7 agonist (300 µg/mouse) or vehicle control (2% DMSO) was administered IV, and experiments depicted in (E-G) performed 1 hour post-administration. (E) Surface availability of receptors glycoprotein Ibα, glycoprotein V (GPV), glycoprotein VI (GPVI), glycoprotein IX (GPIX), αv-integrin, and β3-integrin on murine platelets detected by whole blood flow cytometry (using anti-mouse CD42b/GPIbα-Dylight 649, anti-mouse GPV-FITC, anti-mouse GPVI-FITC, anti-mouse GPIX-FITC, anti-mouse β3-FITC, and anti-mouse αv-FITC). Data in the graphs are presented as mean ± SEM derived from 6 mice per group. (F) Tail bleeding time and (G) plasma coagulation profile (PT, APTT) was evaluated using the START4 platform. Data are mean ± SEM from 5 mice per group. (H) Phosphatidylserine exposure (annexin V-FITC MFI) on human platelets and percentage of annexin V+ platelets under basal resting condition and following CRP (5 µg/mL) stimulation for 1 hour at room temperature in presence/absence of CXCR7 agonist (100 µg/mL)/vehicle control (1% DMSO) given as a pretreatment of 30 minutes at room temperature. Data are mean ± SEM of 4 experiments with healthy donors. I. Thrombin generation under basal condition and in the presence of platelet-activating CRP in platelet-rich plasma (PRP) and platelet-poor plasma (PPP) evaluated by calibrated automated thrombinoscopy (Stago) in presence/absence of CXCR7 agonist (100 µg/mL)/vehicle control (1% DMSO) given as a pretreatment of 30 minutes at room temperature. Data are mean ± SEM of independent experiments performed with n = 4 healthy donors. (H-I) **P < .01, ***P < .001, ****P < .0001 using ANOVA followed by Sidak’s post hoc test. (J) Thromboelastographic analysis (Whole Blood Hemostasis System) using the TEG6s PlateletMapping cartridges (Haemonetics, Germany) evaluating (Ji) the time to initiation of the appearance of first clot (R) and clot strength deciphered as maximum amplitude (MA). (Jii) Percentage of ADP aggregation; percentage of (extent of) inhibition imposed on ADP-induced aggregation, ascertained separately in the HKH (kaolin with heparinase); ActF (ActivatorF); and ADP assays using blood from healthy subjects in presence/absence of CXCR7 agonist (100 µg/mL)/vehicle control (1% DMSO). Data are shown as mean ± SEM of 5 independent experiments. ##P= .002, **P = .003 using a paired Student t test. APC, apocyanin; DMSO, dimethyl sulfoxide; FITC, fluorescein isothiocyanate; MFI, mean fluorescence intensity; PE, phosphatidylethanolamine.
CXCR7 agonist modulates thrombotic and thromboinflammatory platelet response without compromising plasma dependent coagulation. CXCR7 agonist (300 µg/mouse) or vehicle control (2% DMSO) was administered IV 1 hour prior to the surgical procedure. Surface expression of (Ai) ACKR3/CXCR7 (anti-human/mouse CXCR7-PE) and CXCR4 (anti-mouse CXCR4-Fluorescein) on circulating murine platelets. (Aii) Platelet JON/A response (JON/A-PE) and CD62P (anti-mouse CD62P-FITC) surface expression and (Aiii) surface expression of CXCL12/SDF-1α (anti human/mouse CXCL12/SDF-1α- Fluorescein) (denoting degranulation) in basal and CRP-activated (incubated ex vivo for 30 minutes at room temperature) state were evaluated by flow cytometry using blood collected pre-MI (before beginning the surgical procedure) and 24 hours post-MI/RI. (Aiv) Thrombus coverage ex vivo. (B) Levels of proinflammatory mediators in plasma (violin plot with median line) measured with cytometric bead arrays (Legendplex murine 13-plex inflammation panel) 24 hours post-MI/RI. (C) Percentage of CD42b+ (anti-mouse CD42b-FITC)-CD45+ (anti-mouse CD45-APC) platelet-leukocyte aggregates in blood collected pre-MI and 24 hours post-MI/RI. In panels A through C, *P < .05, **P < .01, ***P < .001, ****P < .0001 using Mann-Whitney U test. (Di-Dii) Platelet count and mean platelet volume (MPV) 24 hours post-MI/RI. CXCR7 agonist (300 µg/mouse) or vehicle control (2% DMSO) was administered IV, and experiments depicted in (E-G) performed 1 hour post-administration. (E) Surface availability of receptors glycoprotein Ibα, glycoprotein V (GPV), glycoprotein VI (GPVI), glycoprotein IX (GPIX), αv-integrin, and β3-integrin on murine platelets detected by whole blood flow cytometry (using anti-mouse CD42b/GPIbα-Dylight 649, anti-mouse GPV-FITC, anti-mouse GPVI-FITC, anti-mouse GPIX-FITC, anti-mouse β3-FITC, and anti-mouse αv-FITC). Data in the graphs are presented as mean ± SEM derived from 6 mice per group. (F) Tail bleeding time and (G) plasma coagulation profile (PT, APTT) was evaluated using the START4 platform. Data are mean ± SEM from 5 mice per group. (H) Phosphatidylserine exposure (annexin V-FITC MFI) on human platelets and percentage of annexin V+ platelets under basal resting condition and following CRP (5 µg/mL) stimulation for 1 hour at room temperature in presence/absence of CXCR7 agonist (100 µg/mL)/vehicle control (1% DMSO) given as a pretreatment of 30 minutes at room temperature. Data are mean ± SEM of 4 experiments with healthy donors. I. Thrombin generation under basal condition and in the presence of platelet-activating CRP in platelet-rich plasma (PRP) and platelet-poor plasma (PPP) evaluated by calibrated automated thrombinoscopy (Stago) in presence/absence of CXCR7 agonist (100 µg/mL)/vehicle control (1% DMSO) given as a pretreatment of 30 minutes at room temperature. Data are mean ± SEM of independent experiments performed with n = 4 healthy donors. (H-I) **P < .01, ***P < .001, ****P < .0001 using ANOVA followed by Sidak’s post hoc test. (J) Thromboelastographic analysis (Whole Blood Hemostasis System) using the TEG6s PlateletMapping cartridges (Haemonetics, Germany) evaluating (Ji) the time to initiation of the appearance of first clot (R) and clot strength deciphered as maximum amplitude (MA). (Jii) Percentage of ADP aggregation; percentage of (extent of) inhibition imposed on ADP-induced aggregation, ascertained separately in the HKH (kaolin with heparinase); ActF (ActivatorF); and ADP assays using blood from healthy subjects in presence/absence of CXCR7 agonist (100 µg/mL)/vehicle control (1% DMSO). Data are shown as mean ± SEM of 5 independent experiments. ##P= .002, **P = .003 using a paired Student t test. APC, apocyanin; DMSO, dimethyl sulfoxide; FITC, fluorescein isothiocyanate; MFI, mean fluorescence intensity; PE, phosphatidylethanolamine.
Targeting ACKR3/CXCR7 does not affect basal hemostatic or coagulation response
CXCR7 agonist administration in vivo neither altered bleeding time (Figure 2F) nor plasma coagulation profile (APTT, PT)29 (Figure 2G) of mice. We further verified these observations in human systems in vitro. Phosphatidylserine (PS) exposure on procoagulant platelets support thrombin generation driving coagulation,52 which is severely affected in Scott syndrome, leading to enhanced bleeding.53,54 Increased PS exposure on procoagulant platelets during acute inflammation, as in intensive care unit–admitted COVID-19 patients, correlates with sequential organ failure assessment score and d-dimer levels. COVID-19 patients with thrombotic complications exhibit higher PS externalization.3 Like physiological ligands (CXCL12/SDF1α, MIF),26,27 pharmacological CXCR7 agonist reduced the extent of PS exposure (annexin V mean fluorescence intensity) and percentage of annexin V+ procoagulant platelets (Figure 2H) following CRP stimulation, without affecting basal status. Consequently, CXCR7 agonist counteracted only CRP-induced thrombin generation in PRP without affecting basal thrombin generation in PRP or in PPP (Figure 2I). Thromboelastographic (TEG6s) analysis using PlateletMapping assay confirmed that CXCR7 agonist did not affect clot formation (R) or clot strength (MA) (Figure 2Ji) in kaolin with heparinase activation test or MA in Activator F test. However, it significantly reduced ADP-activated platelet-dependent clot formation (MA), as well as decreased percent of ADP-induced aggregation and increased the extent of aggregation inhibition (Figure 2Jii). Therefore, targeting ACKR3/CXCR7 may regulate activated platelet-driven procoagulant and hemostatic functions without affecting plasma coagulation and basal hemostatic response.
ACKR3/CXCR7 ligation modulates the platelet lipidome
Next, we investigated the molecular mechanisms driving these inhibitory effects. Platelet CXCR4 or ACKR3/CXCR7 surface expression is not altered by activating stimuli but by physiological ligands26,27 and lipoproteins.29 Higher plasma low-density lipoprotein (LDL) levels in CAD patients correspond with significantly increased platelet CXCR4 and relatively enhanced ACKR3/CXCR7 expression (supplemental Figure 6A). Intraplatelet oxidized LDL levels correlate differentially with platelet CXCR4 and ACKR3/CXCR7 surface expression29 in CAD patients, who exhibit a distinctively altered platelet lipidome enriched in atherogenic oxidized phospholipids and ceramides.29 Platelet activatory signaling cascade also engages lipid mediators, while lipid agonists (eg, TxA2) released upon activation substantiate functional response.38,55,56
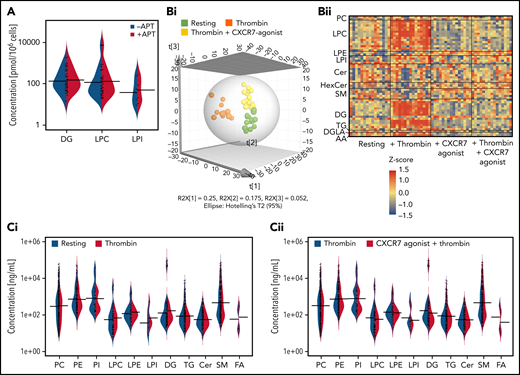
Current lipidomics evaluation in healthy subjects (Figure 3A-F) revealed a significant influence of ACKR3/CXCR7 ligation on the basal platelet lipidome over time, showing significantly reduced levels of several lipid classes including phosphatidylethanolamine, phosphatidylcholine, lysophosphatidylethanolamine (LPE), lysophosphatidylcholine (LPC), and sphingomyelin (Figure 3A). Thrombin-induced activation considerably alters the platelet lipidome.57 Distinct clustering of resting and thrombin-activated platelets (Figure 3B), as well as platelet releasates (Figure 3C), in the presence/absence of CXCR7 agonist in principal components analysis-discriminant analysis (PCA-DA) score plots distinctly demonstrated that ACKR3/CXCR7 ligation may alter basal and counteract thrombin-driven changes to the platelet lipidome in healthy subjects. Thrombin-induced generation (Figure 3D) and release of (Figure 3E) prothrombotic arachidonic acid (AA), 12-hydroxyheptadecatrenoic acid (12-HHT), thromboxane-TxA(B)2 through cyclooxygenase-1 (COX-1), and 12-HETE through 12-lipoxygenase (12-LOX) were reduced upon ACKR3/CXCR7 ligation. 12-HETE promotes platelet activation downstream of FcγRIIA,58 whereas HETE phospholipids promote coagulation in a calcium- and phosphatidylserine-dependent manner.59
![ACKR3/CXCR7 ligation modulates the platelet lipidome: observation from healthy subjects and CAD patients. (A) Washed platelets (200 × 106/sample) from n = 5 healthy donors were treated with CXCR7 agonist (100 µg/mL) for 0, 5, 15, 30, and 60 minutes at room temperature. Heat map showing significant changes in lipid concentration upon treatment with CXCR7 agonist (100 µg/mL) for a period of (0-60) minutes; linear regression model; the significance level for the corrected (false discovery rate correction [FDR]) P values was 0.05. The data were normalized by z-scores for each donor separately and derived from 5 independent experiments performed with 5 healthy donors. Washed platelets (300 × 106/sample) from n = 11 healthy donors were pretreated with CXCR7 agonist (100 µg/mL) or vehicle control (1% DMSO) for 30 minutes at room temperature. Platelets were then either kept under resting condition or activated with thrombin (0.1 U/mL) for 15 minutes at room temperature. The supernatants were separated from the platelet pellets by centrifugation, and both were used for lipid extraction and subsequent lipidomics analysis. PCA-DA score plots showing distinct grouping or clustering of resting platelets, thrombin-activated platelets, CXCR7 agonist plus thrombin-treated platelets, and CXCR7 agonist–treated platelets as analyzed in (B) platelet pellet and (C) platelet releasate/platelet supernatant. Relative abundance reflecting generation of (D) AA, TxA(B)2, 12-HHT, and 12-HETE in thrombin (0.1 U/mL)-activated platelet pellet from n = 11 healthy subjects plus CXCR7 agonist (100 µg/mL) or vehicle control and (E) release of AA, TxA(B)2, 12-HHT, and 12-HETE in thrombin-activated platelet supernatant of healthy subjects plus CXCR7 agonist (100 µg/mL) or vehicle control. Metabolism of (Fi) LPCs, (Fii) diacylglycerol (DGs), and (Fiii) LPIs in thrombin-activated platelets from healthy subjects is significantly reduced by CXCR7 agonist (100 µg/mL) pretreatment with respect to vehicle control. The significance levels for the corrected (FDR) P values are given using nonparametric paired Wilcoxon signed rank test with statistical significance P < .05. Response in the Y-axis of panels D and F refers here to relative signal intensities and were calculated from the raw data (peak heights) after LOWESS normalization. (G) Washed platelets (300 × 106/sample) from CAD patients (Table 2; n = 15 for untargeted analysis of AA and n = 7 for targeted analysis of oxylipins) were pretreated with CXCR7 agonist (100 µg/mL) or vehicle control (1% DMSO) for 30 minutes at room temperature. Platelets were then activated with thrombin (0.1 U/mL) for 15 minutes at room temperature. Generation of platelet-activating prothrombotic lipid mediators AA, TxA(B)2, 12-HHT, and 12-HETE in thrombin (0.1 U/mL)-activated platelets was significantly reduced upon ex vivo pretreatment with CXCR7 agonist (100 µg/mL) with respect to vehicle control. The significance levels for the corrected (FDR) P values are given using nonparametric paired Wilcoxon signed rank test with statistical significance P < .05. DMSO, dimethyl sulfoxide; LOWESS, locally weighted scatterplot smoothing; PCA-DA, principal components analysis-discriminant analysis.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/11/10.1182_blood.2021013097/5/m_bloodbld2021013097f3.png?Expires=1765892644&Signature=jIRnWvOIXowJbF9dqmkfyRsip8Pbl~PySKvUkwNByzcFM3qnXbNn5-nkWaKIQ7FDrZCBBjcdlFTW9XBl2E~pPQc7F5~2bK5qHwxwlVmXq-iQf1slHt50XrU05ecPgaVQuc3wKrPkPqdlfWjzjUMr67oZ-6ubq2OaYgYv51JY~YpLVcuudY3cWe51G8A2PWD3oaHWVzMRZJumVb6V~QC3RGIdiuqWScJ0nNnz9zOiz8TFE0avw5sod2qZCqS7rOi1Kv2b91Y3U-M4UYM8W~8dqUafx2HN0ewub-XBQpViSGddPYOuGPbZsMl8Q8jqAHBtePvvilJPtYgicuhQGRdapQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
ACKR3/CXCR7 ligation modulates the platelet lipidome: observation from healthy subjects and CAD patients. (A) Washed platelets (200 × 106/sample) from n = 5 healthy donors were treated with CXCR7 agonist (100 µg/mL) for 0, 5, 15, 30, and 60 minutes at room temperature. Heat map showing significant changes in lipid concentration upon treatment with CXCR7 agonist (100 µg/mL) for a period of (0-60) minutes; linear regression model; the significance level for the corrected (false discovery rate correction [FDR]) P values was 0.05. The data were normalized by z-scores for each donor separately and derived from 5 independent experiments performed with 5 healthy donors. Washed platelets (300 × 106/sample) from n = 11 healthy donors were pretreated with CXCR7 agonist (100 µg/mL) or vehicle control (1% DMSO) for 30 minutes at room temperature. Platelets were then either kept under resting condition or activated with thrombin (0.1 U/mL) for 15 minutes at room temperature. The supernatants were separated from the platelet pellets by centrifugation, and both were used for lipid extraction and subsequent lipidomics analysis. PCA-DA score plots showing distinct grouping or clustering of resting platelets, thrombin-activated platelets, CXCR7 agonist plus thrombin-treated platelets, and CXCR7 agonist–treated platelets as analyzed in (B) platelet pellet and (C) platelet releasate/platelet supernatant. Relative abundance reflecting generation of (D) AA, TxA(B)2, 12-HHT, and 12-HETE in thrombin (0.1 U/mL)-activated platelet pellet from n = 11 healthy subjects plus CXCR7 agonist (100 µg/mL) or vehicle control and (E) release of AA, TxA(B)2, 12-HHT, and 12-HETE in thrombin-activated platelet supernatant of healthy subjects plus CXCR7 agonist (100 µg/mL) or vehicle control. Metabolism of (Fi) LPCs, (Fii) diacylglycerol (DGs), and (Fiii) LPIs in thrombin-activated platelets from healthy subjects is significantly reduced by CXCR7 agonist (100 µg/mL) pretreatment with respect to vehicle control. The significance levels for the corrected (FDR) P values are given using nonparametric paired Wilcoxon signed rank test with statistical significance P < .05. Response in the Y-axis of panels D and F refers here to relative signal intensities and were calculated from the raw data (peak heights) after LOWESS normalization. (G) Washed platelets (300 × 106/sample) from CAD patients (Table 2; n = 15 for untargeted analysis of AA and n = 7 for targeted analysis of oxylipins) were pretreated with CXCR7 agonist (100 µg/mL) or vehicle control (1% DMSO) for 30 minutes at room temperature. Platelets were then activated with thrombin (0.1 U/mL) for 15 minutes at room temperature. Generation of platelet-activating prothrombotic lipid mediators AA, TxA(B)2, 12-HHT, and 12-HETE in thrombin (0.1 U/mL)-activated platelets was significantly reduced upon ex vivo pretreatment with CXCR7 agonist (100 µg/mL) with respect to vehicle control. The significance levels for the corrected (FDR) P values are given using nonparametric paired Wilcoxon signed rank test with statistical significance P < .05. DMSO, dimethyl sulfoxide; LOWESS, locally weighted scatterplot smoothing; PCA-DA, principal components analysis-discriminant analysis.
ACKR3/CXCR7 ligation modulates the platelet lipidome: observation from healthy subjects and CAD patients. (A) Washed platelets (200 × 106/sample) from n = 5 healthy donors were treated with CXCR7 agonist (100 µg/mL) for 0, 5, 15, 30, and 60 minutes at room temperature. Heat map showing significant changes in lipid concentration upon treatment with CXCR7 agonist (100 µg/mL) for a period of (0-60) minutes; linear regression model; the significance level for the corrected (false discovery rate correction [FDR]) P values was 0.05. The data were normalized by z-scores for each donor separately and derived from 5 independent experiments performed with 5 healthy donors. Washed platelets (300 × 106/sample) from n = 11 healthy donors were pretreated with CXCR7 agonist (100 µg/mL) or vehicle control (1% DMSO) for 30 minutes at room temperature. Platelets were then either kept under resting condition or activated with thrombin (0.1 U/mL) for 15 minutes at room temperature. The supernatants were separated from the platelet pellets by centrifugation, and both were used for lipid extraction and subsequent lipidomics analysis. PCA-DA score plots showing distinct grouping or clustering of resting platelets, thrombin-activated platelets, CXCR7 agonist plus thrombin-treated platelets, and CXCR7 agonist–treated platelets as analyzed in (B) platelet pellet and (C) platelet releasate/platelet supernatant. Relative abundance reflecting generation of (D) AA, TxA(B)2, 12-HHT, and 12-HETE in thrombin (0.1 U/mL)-activated platelet pellet from n = 11 healthy subjects plus CXCR7 agonist (100 µg/mL) or vehicle control and (E) release of AA, TxA(B)2, 12-HHT, and 12-HETE in thrombin-activated platelet supernatant of healthy subjects plus CXCR7 agonist (100 µg/mL) or vehicle control. Metabolism of (Fi) LPCs, (Fii) diacylglycerol (DGs), and (Fiii) LPIs in thrombin-activated platelets from healthy subjects is significantly reduced by CXCR7 agonist (100 µg/mL) pretreatment with respect to vehicle control. The significance levels for the corrected (FDR) P values are given using nonparametric paired Wilcoxon signed rank test with statistical significance P < .05. Response in the Y-axis of panels D and F refers here to relative signal intensities and were calculated from the raw data (peak heights) after LOWESS normalization. (G) Washed platelets (300 × 106/sample) from CAD patients (Table 2; n = 15 for untargeted analysis of AA and n = 7 for targeted analysis of oxylipins) were pretreated with CXCR7 agonist (100 µg/mL) or vehicle control (1% DMSO) for 30 minutes at room temperature. Platelets were then activated with thrombin (0.1 U/mL) for 15 minutes at room temperature. Generation of platelet-activating prothrombotic lipid mediators AA, TxA(B)2, 12-HHT, and 12-HETE in thrombin (0.1 U/mL)-activated platelets was significantly reduced upon ex vivo pretreatment with CXCR7 agonist (100 µg/mL) with respect to vehicle control. The significance levels for the corrected (FDR) P values are given using nonparametric paired Wilcoxon signed rank test with statistical significance P < .05. DMSO, dimethyl sulfoxide; LOWESS, locally weighted scatterplot smoothing; PCA-DA, principal components analysis-discriminant analysis.
Platelets from CAD patients show increased levels of phospholipase-A2 (LPC, lyophosphatidylinositol [LPI]) and phospholipase-C (diacylglycerol [DG]) metabolites.29 LPCs, a major constituent of platelet-derived microvesicles,60 induce platelet activation and platelet-monocyte aggregate formation. LPCs influence plaque instability and are richly deposited at unstable areas of atherosclerotic plaques60 that may support atherothrombosis. CXCR7 agonist reduced generation of LPCs (Figure 3Fi), DGs (Figure 3Fii), and LPIs (Figure 3Fiii) in thrombin-activated platelets from healthy subjects. This was a unique regulatory influence of a chemokine receptor (ACKR3/CXCR7) on the generation of prothrombotic (eg, LPI, DG, TxA2, 12-HETE) and atherogenic (eg, LPC) lipids in platelets, which may deliver them at the site of vascular inflammation/injury during atheroprogression. As in platelets from healthy donors, CXCR7 agonist treatment ex vivo significantly counteracted subsequent thrombin-induced generation of prothrombotic lipid mediators (AA, 12-HHT, TxA(B)2, 12-HETE) in platelets from CAD patients (Table 2; Figure 3G), who also showed significantly increased platelet ACKR3/CXCR7 surface expression as compared to healthy subjects (supplemental Figure 6B).
Baseline characteristics of CAD patients enrolled for the analysis of platelet lipidome and functions following ex vivo treatment with CXCR7 agonist
| CAD patient characteristics | CAD (n=20) |
|---|---|
| Age (mean ± SD) | 71.9 (± 11.2) |
| Male gender | 14 (70%) |
| LVEF% at admission (mean ± SD) | 51.7 (± 7.8) |
| Cardiovascular risk factors | |
| Arterial hypertension | 18 (90%) |
| Hyperlipidemia | 11 (55%) |
| Diabetes mellitus type 2 | 9 (45%) |
| Smoking | 4 (20%) |
| Medication on admission | |
| ASA | 9 (45%) |
| Clopidogrel | 3 (15%) |
| Prasugrel | 0 (0%) |
| Ticagrelor | 0 (0%) |
| ACE inhibitors | 7 (35%) |
| ARBs | 7 (35%) |
| β blockers | 10 (50%) |
| Ca-channel inhibitors | 6 (30%) |
| Diuretics | 6 (30%) |
| Statins | 11(55%) |
| Reason of admission/clinical diagnosis | |
| ACS | 4 (20%) |
| CCS | 16 (80%) |
| Plasma lipid profile | |
| Total cholesterol | 170.1 (± 59.8) |
| LDL cholesterol | 109.3 (± 49.9) |
| HDL cholesterol | 47.8 (± 17.2) |
| Triglycerides | 134.6 (± 91) |
| CAD patient characteristics | CAD (n=20) |
|---|---|
| Age (mean ± SD) | 71.9 (± 11.2) |
| Male gender | 14 (70%) |
| LVEF% at admission (mean ± SD) | 51.7 (± 7.8) |
| Cardiovascular risk factors | |
| Arterial hypertension | 18 (90%) |
| Hyperlipidemia | 11 (55%) |
| Diabetes mellitus type 2 | 9 (45%) |
| Smoking | 4 (20%) |
| Medication on admission | |
| ASA | 9 (45%) |
| Clopidogrel | 3 (15%) |
| Prasugrel | 0 (0%) |
| Ticagrelor | 0 (0%) |
| ACE inhibitors | 7 (35%) |
| ARBs | 7 (35%) |
| β blockers | 10 (50%) |
| Ca-channel inhibitors | 6 (30%) |
| Diuretics | 6 (30%) |
| Statins | 11(55%) |
| Reason of admission/clinical diagnosis | |
| ACS | 4 (20%) |
| CCS | 16 (80%) |
| Plasma lipid profile | |
| Total cholesterol | 170.1 (± 59.8) |
| LDL cholesterol | 109.3 (± 49.9) |
| HDL cholesterol | 47.8 (± 17.2) |
| Triglycerides | 134.6 (± 91) |
Arterial blood was collected from CAD (n = 20) patients during the percutaneous coronary intervention (PCI) procedure as previously described.70 Administration of antiplatelet therapy and statin were considered from the record of medications upon admission.
By comparison, levels of DGs, LPCs, and LPIs (Figure 4A; supplemental Figure 6Ci-Cv) in platelets from CAD patients (n = 107; Table 3) were not significantly regulated by APT, even in combination with statin (68.2% patients) (Figure 4A; supplemental Excel File 1). Although ASA inhibits COX-1–metabolized TxA2 generation, it might not influence generation of upstream phospholipase metabolites (eg, LPCs), warranting alternate strategies to modulate the atherothrombotic platelet lipidome. Thrombin-induced ex vivo changes to the platelet lipidome was observed in CAD patients (n = 15) (Figure 4Bi-Bii) despite 5 out of 15 patients being on ASA. Treatment of platelets from these CAD patients (Table 2) with CXCR7 agonist ex vivo significantly counteracted subsequent thrombin-induced changes to the platelet lipidome (Figure 4Bi-Bii), particularly atherogenic LPCs, lipid mediator DGs, (Figure 4C-D), and to a certain extent, LPIs (Figure 4Diii). Combined with its effects on lipid agonists (AA, TxA2, 12-HETE) (Figure 3G), this may account for the antithrombotic effects of CXCR7 agonist.
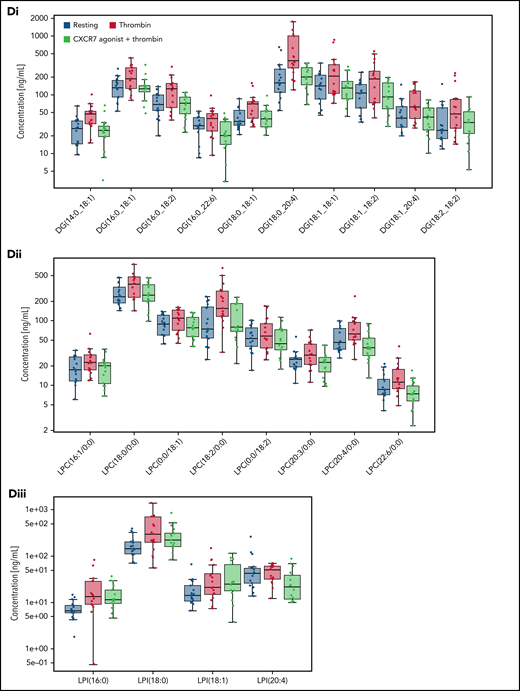
Effect of antiplatelet therapy in clinical practice and ex vivo CXCR7 agonist treatment on the platelet lipidome of CAD patients. Data are derived from untargeted lipidomics analysis of platelets. The asymmetric beanplot in (A) is derived from data on the basal platelet lipidome of CAD (n = 107, Table 3) patients. It represents the distribution of the concentration levels of individual lipid species within each of the lipid classes DG, LPC, and LPI among CAD patients treated with antiplatelet therapy (APT) (n = 46) and those without APT (n = 61). Smaller lines indicate concentration levels of individual lipid species within a group; the larger lines mark the average concentration of all detected lipid species within each class and group. (Bi-Diii) Lipidomics analysis of platelets from n = 15 CAD patients (Table 2) in their resting status and after thrombin (0.1 U/mL)-induced activation for 15 minutes at room temperature in presence/absence of ex vivo pretreatment with CXCR7 agonist (100 µg/mL) or vehicle control (1% DMSO) for 30 minutes at room temperature. Lipid concentrations were scaled for each donor by a z-score calculation. The 3D representation of a PLS-DA model in panel Bi shows clear clustering of platelet samples in accordance with their resting status, thrombin-induced activation, and thrombin plus CXCR7 agonist treatment conditions (six components with R2X(cum) = 0.566, R2Y(cum) = 0.971, and Q2(cum) = 0.780). The heat map in panel Bii is based on lipids with a variable importance in projection (VIP) score (which is a measure of a variable’s importance in the PLS-DA model) >1.5 in the PLS-DA model further illustrates alterations in the lipidome, especially in thrombin-activated platelets. (C) Asymmetric beanplots indicate changes in the platelet lipidome for the different lipid classes phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylinositol (PI), LPC, lysophosphatidylethanolamine (LPE), LPI, DG, triacylglycerol (TG), ceramides (Cer), sphingomyelin (SM), and fatty acids (FA), either comparing (Ci) resting vs thrombin-activated platelets or (Cii) thrombin-activated vs thrombin plus CXCR7 agonist–treated platelets (small lines denote individual lipid species and larger markings denote average concentration per lipid class and group). (Di-Dii) Boxplots depict changing concentration levels of significantly altered lipids in thrombin vs thrombin plus CXCR7 agonist–treated platelets (nonparametric paired Wilcoxon signed rank test with FDR correction, significance level: q-value < 0.05), which belong to the DG and LPC lipid classes. (Diii) The boxplots of 4 LPI species (showing a trend of reduced generation in the presence of CXCR7 agonist as compared with thrombin alone, without reaching statistical significance). All lipid concentrations were determined by relative quantification using class-specific isotope labeled internal standard (ILIS) related to 100 µL lipid extract from 3 × 108 platelets. DMSO, dimethyl sulfoxide; PLS-DA, partial least square-discriminant analysis.
Effect of antiplatelet therapy in clinical practice and ex vivo CXCR7 agonist treatment on the platelet lipidome of CAD patients. Data are derived from untargeted lipidomics analysis of platelets. The asymmetric beanplot in (A) is derived from data on the basal platelet lipidome of CAD (n = 107, Table 3) patients. It represents the distribution of the concentration levels of individual lipid species within each of the lipid classes DG, LPC, and LPI among CAD patients treated with antiplatelet therapy (APT) (n = 46) and those without APT (n = 61). Smaller lines indicate concentration levels of individual lipid species within a group; the larger lines mark the average concentration of all detected lipid species within each class and group. (Bi-Diii) Lipidomics analysis of platelets from n = 15 CAD patients (Table 2) in their resting status and after thrombin (0.1 U/mL)-induced activation for 15 minutes at room temperature in presence/absence of ex vivo pretreatment with CXCR7 agonist (100 µg/mL) or vehicle control (1% DMSO) for 30 minutes at room temperature. Lipid concentrations were scaled for each donor by a z-score calculation. The 3D representation of a PLS-DA model in panel Bi shows clear clustering of platelet samples in accordance with their resting status, thrombin-induced activation, and thrombin plus CXCR7 agonist treatment conditions (six components with R2X(cum) = 0.566, R2Y(cum) = 0.971, and Q2(cum) = 0.780). The heat map in panel Bii is based on lipids with a variable importance in projection (VIP) score (which is a measure of a variable’s importance in the PLS-DA model) >1.5 in the PLS-DA model further illustrates alterations in the lipidome, especially in thrombin-activated platelets. (C) Asymmetric beanplots indicate changes in the platelet lipidome for the different lipid classes phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylinositol (PI), LPC, lysophosphatidylethanolamine (LPE), LPI, DG, triacylglycerol (TG), ceramides (Cer), sphingomyelin (SM), and fatty acids (FA), either comparing (Ci) resting vs thrombin-activated platelets or (Cii) thrombin-activated vs thrombin plus CXCR7 agonist–treated platelets (small lines denote individual lipid species and larger markings denote average concentration per lipid class and group). (Di-Dii) Boxplots depict changing concentration levels of significantly altered lipids in thrombin vs thrombin plus CXCR7 agonist–treated platelets (nonparametric paired Wilcoxon signed rank test with FDR correction, significance level: q-value < 0.05), which belong to the DG and LPC lipid classes. (Diii) The boxplots of 4 LPI species (showing a trend of reduced generation in the presence of CXCR7 agonist as compared with thrombin alone, without reaching statistical significance). All lipid concentrations were determined by relative quantification using class-specific isotope labeled internal standard (ILIS) related to 100 µL lipid extract from 3 × 108 platelets. DMSO, dimethyl sulfoxide; PLS-DA, partial least square-discriminant analysis.
Baseline characteristics of CAD patients enrolled for analysis of the basal platelet lipidome
| CAD patient characteristics | CAD (n = 107) |
|---|---|
| Age (mean ± SD) | 71.2 (± 10.7) |
| Male gender | 79 (73.8%) |
| LVEF% at admission (mean ± SD) | 51.9 (± 11.2) |
| Cardiovascular risk factors | |
| Arterial hypertension | 86 (80.4%) |
| Hyperlipidemia | 52 (48.6%) |
| Diabetes mellitus type 2 | 31 (29.0%) |
| Smoking | 19 (17.8%) |
| Medication on admission | |
| ASA | 54 (50.5%) |
| Clopidogrel | 12 (11.2%) |
| Prasugrel | 4 (3.7%) |
| Ticagrelor | 12 (11.2%) |
| ACE inhibitors | 56 (52.3%) |
| ARBs | 26 (24.3%) |
| β blockers | 73 (68.2%) |
| Statins | 73 (68.2%) |
| Reason of admission/clinical diagnosis | |
| ACS | 26 (24.3%) |
| CCS | 81 (75.7%) |
| Plasma lipid profile | |
| Total cholesterol (mg/dL) | 161.3 (± 45.4) |
| LDL cholesterol (mg/dL) | 96.2 (± 34.4) |
| HDL cholesterol (mg/dL) | 48.6 (± 17.4) |
| Triglycerides (mg/dL) | 145.3 (± 84.5) |
| CAD patient characteristics | CAD (n = 107) |
|---|---|
| Age (mean ± SD) | 71.2 (± 10.7) |
| Male gender | 79 (73.8%) |
| LVEF% at admission (mean ± SD) | 51.9 (± 11.2) |
| Cardiovascular risk factors | |
| Arterial hypertension | 86 (80.4%) |
| Hyperlipidemia | 52 (48.6%) |
| Diabetes mellitus type 2 | 31 (29.0%) |
| Smoking | 19 (17.8%) |
| Medication on admission | |
| ASA | 54 (50.5%) |
| Clopidogrel | 12 (11.2%) |
| Prasugrel | 4 (3.7%) |
| Ticagrelor | 12 (11.2%) |
| ACE inhibitors | 56 (52.3%) |
| ARBs | 26 (24.3%) |
| β blockers | 73 (68.2%) |
| Statins | 73 (68.2%) |
| Reason of admission/clinical diagnosis | |
| ACS | 26 (24.3%) |
| CCS | 81 (75.7%) |
| Plasma lipid profile | |
| Total cholesterol (mg/dL) | 161.3 (± 45.4) |
| LDL cholesterol (mg/dL) | 96.2 (± 34.4) |
| HDL cholesterol (mg/dL) | 48.6 (± 17.4) |
| Triglycerides (mg/dL) | 145.3 (± 84.5) |
CAD (n = 107) patients were enrolled separately for lipidomic analysis of washed platelets isolated from arterial blood collected during percutaneous coronary intervention (PCI) procedure.70 Administration of antiplatelet therapy and statin were considered from the record of medications upon admission.
Reduced generation of lipid mediators affects platelet activatory signaling cascade and functions
Signaling events triggered by external stimuli at the membrane receptors (TRAP/thrombin-PARs, collagen/CRP-xl-GPVI, ADP-P2Y12) converge downstream at the level of phospholipases.38,55 Because ACKR3/CXCR7 ligation significantly altered the lipidome of both resting and activated platelets, we assessed some prime components of the platelet activatory signaling cascade. CXCR7 agonist reduced CRP-GPVI–induced phosphorylation/activation of PLCγ and Src family kinases (Figure 5A). A decrease in thrombin-induced LPI generation (Figure 3Fiii) corroborated with reduced intracellular calcium mobilization in the presence of CXCR7 agonist (Figure 5B), whereas reduced PLCγ activation, and consequently DG production (Figure 3Fii), corresponded with compromised PKC activation following CRP-GPVI stimulation (Figure 5C). CRP-GPVI–induced phosphorylation of PI3K, Akt, and p38MAPK in the activatory signaling network were also affected (Figure 5C).
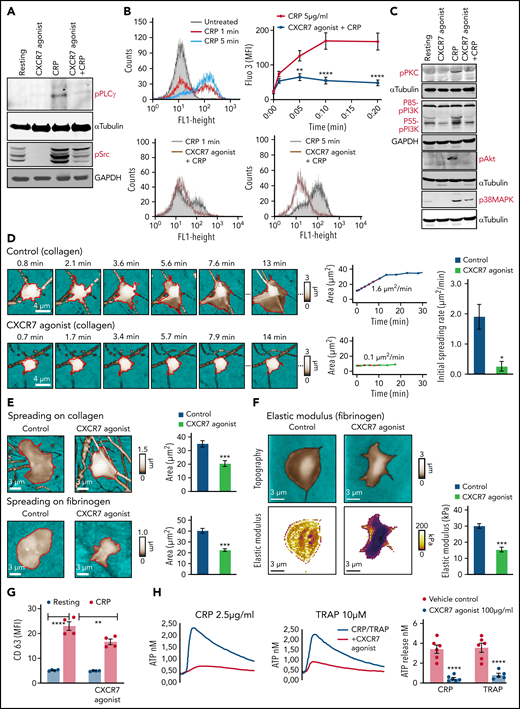
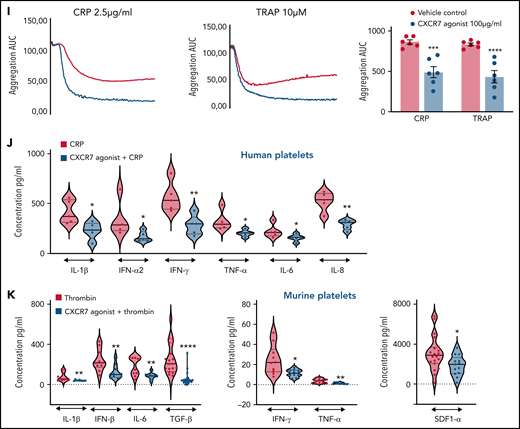
CXCR7 agonist–induced changes in platelet lipidome affect activatory mediators and functional response. (A) Western blot analysis showing phosphorylation of PLCγ and Src family kinases in response to CRP (5 µg/mL) stimulation for 10 minutes at room temperature in presence/absence of CXCR7 agonist (100 µg/mL) or vehicle control (1% DMSO) given as a pretreatment of 15 minutes at room temperature. (B) Flow cytometric histogram overlays and data showing CRP (5 µg/mL)-induced intraplatelet calcium mobilization detected with Fluo-3 am (5 µM) in presence/absence of CXCR7 agonist (100 µg/mL)/vehicle control in whole blood assay gating for platelet-specific marker CD42b (anti-human CD42b-PE). Data are mean ± SEM from 5 experiments; **P < .0077, ****P < .0001 vs CRP using ANOVA followed by Bonferroni’s multiple comparison test. (C) Phosphorylation of PKC, PI3K, Akt, and p38MAPK triggered by CRP (5 µg/mL) in presence/absence of CXCR7 agonist (100 µg/mL)/vehicle control (1% DMSO). In panels A and C, data are representative of 3 western blots. (D) SICM imaging showed reduced initial spreading rate on collagen (100 µg/mL)-coated surfaces in presence/absence of CXCR7 agonist (50 µg/mL) or vehicle control (0.1% DMSO). (E) Decreased final spreading area of CXCR7 agonist (50 µg/mL)-treated platelets on collagen- and fibrinogen (100 µg/mL)-coated surfaces with respect to vehicle control. Data are mean ± SEM from 3 experiments. Student t test; *P < .05, ***P < .001. (F) SICM stiffness mapping showing reduced elastic modulus of platelets pretreated with CXCR7 agonist (50 µg/mL) while interacting with fibrinogen-coated surfaces. Plots show geometric mean ± SEM from 3 experiments. Student t test, ***P < .001. Number of platelets: 5 to 6 in panel D, 18 to 24 in panel E, and 25 to 31 in panel F. (G) Surface expression of CD63 (δ-granule release, anti-human CD63-FITC) detected by flow cytometry following CRP (5 µg/mL)-induced activation for 30 minutes at room temperature in presence/absence of CXCR7 agonist (100 µg/mL)/vehicle control (1% DMSO) given as a pretreatment of 30 minutes at room temperature. Data are mean ± SEM from 4 experiments; **P < .002, ****P < .0001 using ANOVA followed by Sidak’s multiple comparison text. (H) CRP (2.5 µg/mL) and TRAP (10 µM) induced release of ATP from δ-granules in presence/absence of CXCR7 agonist (100 µg/mL)/vehicle control ascertained by lumi-aggregometry. Extracellular ATP released from activated platelets was assessed employing the luciferase bioluminescent assay, and the amount of ATP release was calculated using the exogenously added ATP standard with aggrolink-8 software (ChronoLog). (I) Corresponding aggregatory response to CRP (2.5 µg/mL) and TRAP (10 µM) for a duration of 10 minutes at 37°C and under a stirring speed of 1000 revolutions per minute (RPM) in presence/absence of CXCR7 agonist (100 µg/mL)/vehicle control. In panels H through I, data are mean ± SEM from 6 experiments with healthy donors; ***P = .0002, ****P < .0001 using ANOVA followed by Sidak’s multiple comparison text. Washed human (200 × 106/sample) and murine (100 × 106/sample) were preincubated with CXCR7 agonist (100 µg/mL) or vehicle control (1% DMSO) for 30 minutes and thereafter stimulated with platelet agonists as specified below for 30 and 15 minutes, respectively, at room temperature. Samples were then centrifuged to collect activated platelet supernatant. Violin plots (line denotes median) showing release of inflammatory mediators from (J) CRP (5 µg/mL)-activated human (Legendplex human 13-plex inflammation panel) and (K) thrombin (0.1 U/mL)-activated murine platelets (Legendplex murine 13-plex inflammation panel) evaluated by cytometric bead arrays. In panels J and K, *P < .05, **P < .01, ****P < .0001 using Mann-Whitney U test for each analyte. Data are plotted as mean ± SEM from 5 healthy donors in panel J and from 10 mice in panel K. DMSO, dimethyl sulfoxide.
CXCR7 agonist–induced changes in platelet lipidome affect activatory mediators and functional response. (A) Western blot analysis showing phosphorylation of PLCγ and Src family kinases in response to CRP (5 µg/mL) stimulation for 10 minutes at room temperature in presence/absence of CXCR7 agonist (100 µg/mL) or vehicle control (1% DMSO) given as a pretreatment of 15 minutes at room temperature. (B) Flow cytometric histogram overlays and data showing CRP (5 µg/mL)-induced intraplatelet calcium mobilization detected with Fluo-3 am (5 µM) in presence/absence of CXCR7 agonist (100 µg/mL)/vehicle control in whole blood assay gating for platelet-specific marker CD42b (anti-human CD42b-PE). Data are mean ± SEM from 5 experiments; **P < .0077, ****P < .0001 vs CRP using ANOVA followed by Bonferroni’s multiple comparison test. (C) Phosphorylation of PKC, PI3K, Akt, and p38MAPK triggered by CRP (5 µg/mL) in presence/absence of CXCR7 agonist (100 µg/mL)/vehicle control (1% DMSO). In panels A and C, data are representative of 3 western blots. (D) SICM imaging showed reduced initial spreading rate on collagen (100 µg/mL)-coated surfaces in presence/absence of CXCR7 agonist (50 µg/mL) or vehicle control (0.1% DMSO). (E) Decreased final spreading area of CXCR7 agonist (50 µg/mL)-treated platelets on collagen- and fibrinogen (100 µg/mL)-coated surfaces with respect to vehicle control. Data are mean ± SEM from 3 experiments. Student t test; *P < .05, ***P < .001. (F) SICM stiffness mapping showing reduced elastic modulus of platelets pretreated with CXCR7 agonist (50 µg/mL) while interacting with fibrinogen-coated surfaces. Plots show geometric mean ± SEM from 3 experiments. Student t test, ***P < .001. Number of platelets: 5 to 6 in panel D, 18 to 24 in panel E, and 25 to 31 in panel F. (G) Surface expression of CD63 (δ-granule release, anti-human CD63-FITC) detected by flow cytometry following CRP (5 µg/mL)-induced activation for 30 minutes at room temperature in presence/absence of CXCR7 agonist (100 µg/mL)/vehicle control (1% DMSO) given as a pretreatment of 30 minutes at room temperature. Data are mean ± SEM from 4 experiments; **P < .002, ****P < .0001 using ANOVA followed by Sidak’s multiple comparison text. (H) CRP (2.5 µg/mL) and TRAP (10 µM) induced release of ATP from δ-granules in presence/absence of CXCR7 agonist (100 µg/mL)/vehicle control ascertained by lumi-aggregometry. Extracellular ATP released from activated platelets was assessed employing the luciferase bioluminescent assay, and the amount of ATP release was calculated using the exogenously added ATP standard with aggrolink-8 software (ChronoLog). (I) Corresponding aggregatory response to CRP (2.5 µg/mL) and TRAP (10 µM) for a duration of 10 minutes at 37°C and under a stirring speed of 1000 revolutions per minute (RPM) in presence/absence of CXCR7 agonist (100 µg/mL)/vehicle control. In panels H through I, data are mean ± SEM from 6 experiments with healthy donors; ***P = .0002, ****P < .0001 using ANOVA followed by Sidak’s multiple comparison text. Washed human (200 × 106/sample) and murine (100 × 106/sample) were preincubated with CXCR7 agonist (100 µg/mL) or vehicle control (1% DMSO) for 30 minutes and thereafter stimulated with platelet agonists as specified below for 30 and 15 minutes, respectively, at room temperature. Samples were then centrifuged to collect activated platelet supernatant. Violin plots (line denotes median) showing release of inflammatory mediators from (J) CRP (5 µg/mL)-activated human (Legendplex human 13-plex inflammation panel) and (K) thrombin (0.1 U/mL)-activated murine platelets (Legendplex murine 13-plex inflammation panel) evaluated by cytometric bead arrays. In panels J and K, *P < .05, **P < .01, ****P < .0001 using Mann-Whitney U test for each analyte. Data are plotted as mean ± SEM from 5 healthy donors in panel J and from 10 mice in panel K. DMSO, dimethyl sulfoxide.
By influencing the lipidome, ACKR3/CXCR7 ligation may modulate the activatory signaling cascade that mediates platelet responsiveness to external stimuli (eg, interaction with physiological matrices, aggregation, and thromboinflammatory secretion). Live imaging of single platelet response by SICM demonstrated that CXCR7 agonist pretreatment affected adhesion to collagen and fibrinogen (Figure 5D-E). Recording of dynamic platelet response showed reduced initial spreading rate on collagen (Figure 5D) and a significantly decreased final spreading area on collagen and fibrinogen (Figure 5E). Although CXCR7 agonist administration did not alter the surface availability of receptors (Figure 2E), a regulation on receptor-mediated signaling events affected platelet interaction with physiological matrices. A gradual increase in cellular stiffness accompanies platelet adhesion and spreading.39 The elastic modulus (“stiffness”) of platelets adhering to fibrinogen was also reduced (Figure 5F) by CXCR7 agonist pretreatment, in accordance with its impact on platelet adhesion and spreading.
Similar to its effect on CD62P, CXCR7 agonist reduced CD63 surface expression (Figure 5G), and ATP release from δ-granules (Figure 5H) of CRP (GPVI)- and TRAP (PAR1)-activated platelets. Combined with reduced release of lipid agonists (eg, TxA2) (Figure 3), it affected aggregation response to CRP and TRAP (Figure 5I). CXCR7 agonist also reduced degranulation of inflammatory cytokines and chemokines in vitro from α-granules of CRP-activated human (Figure 5J) and thrombin-activated murine platelets (Figure 5K) that mediate atheroprogression14 and contribute (eg, IL-1β, IL-6, IFN-γ) to plasma levels in acute thromboinflammatory conditions.6
Favoring generation of antiplatelet lipids, ACKR3/CXCR7 ligation triggers negative modulators of platelet response
We also explored the influence of ACKR3/CXCR7 ligation on platelet inhibitory signaling. Although ACKR3/CXCR7 is a noncanonical GPCR,32 we observed an unexpected increase in cAMP (Figure 6A), in contrast to Gαi-coupled CXCR4, which decreases cAMP levels to facilitate thrombotic functions.30 Moreover, G-protein coupling antagonist peptide and Gαs-coupling inhibitor (melittin) counteracted the inhibitory effects of CXCR7 agonist on CRP-GPVI–induced degranulation, integrin activation (supplemental Figure 7Ai-Aii), collagen-induced aggregation (supplemental Figure 7Aiii), and thrombus formation (supplemental Figure 7Aiv) without affecting basal response to activating stimuli (supplemental Figure 7Ai-Aiv). However, in case of melittin, in addition to preventing Gαs coupling, a stimulatory effect on Gαi61 and inhibitory effect on adenylyl cyclase62 could also have contributed to this counteracting influence. Neither G-protein coupling antagonist nor melittin triggered release of lipid mediators (AA, TxA2, 12-HHT, 12-HETE, 12-HETrE) (supplemental Figure 7Bi) or altered the platelet lipidome to a significant extent (supplemental Figure 7Bii).
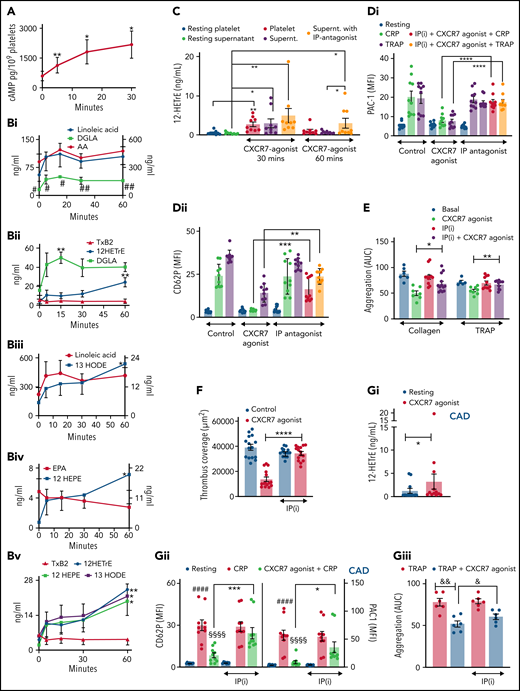
CXCR7 agonist–induced generation of antiplatelet lipid 12-HETrE and platelet inhibitory mediator cAMP. Washed platelets (200 × 106/sample) from n = 5 healthy donors were treated with CXCR7 agonist (100 µg/mL) for 0, 5, 15, and 30 minutes at room temperature. Thereafter platelet pellets were collected by centrifugation and used for detection of cAMP levels by LC-ESI-MS/MS analysis. (A) Gradual elevation in intraplatelet cAMP in response to CXCR7 agonist (100 µg/mL); *P < .05, **P < .01 vs 0 minute by Wilcoxon matched-pairs signed rank test, n = 5 donors. Washed human platelets (200 × 106/sample) from n = 5 healthy donors were treated with CXCR7 agonist (100 µg/mL) for 0, 5, 15, 30, and 60 minutes at room temperature. Intraplatelet levels of (Bi) linoleic acid (right y-axis), AA, and DGLA (left y-axis); (Bii) DGLA, 12-HETrE, and TxA(B)2; (Biii) linoleic acid (left y-axis) and 13-HODE (right y-axis); (Biv) EPA (left y-axis) and 12-HEPE (right y-axis); and (Bv) 12-HETrE, 12-HEPE, and 13-HODE as compared with TxA(B)2. #P < .05 and ##P < .01 vs AA; *P < .05, **P < .01 vs 0 minute using Welch’s t test. (C) Washed platelets (300 × 106/sample) from n = 9 healthy donors were treated with CXCR7 agonist (100 µg/mL) or vehicle control (1% DMSO) for 30 and 60 minutes at room temperature. IP receptor antagonist (RO-1138452 10 µM) was given as a pretreatment before CXCR7 agonist for 15 minutes at room temperature. At the end of incubation period, the supernatants were separated from the platelet pellets by centrifugation, and both were used for lipid extraction and subsequent targeted lipidomics analysis for 12-HETrE. Data are mean ± SEM. The significance levels for the corrected (FDR) P values are given using nonparametric paired Wilcoxon signed rank test with statistical significance *P < .05, **P < .01. Experiments done with blood from healthy donors (n = 5) showing inhibitory effect of CXCR7 agonist (100 µg/mL) on CRP (5 µg/mL) and TRAP (25 µM)-induced platelet (Di) PAC-1 binding, (Dii) CD62P surface expression (flow cytometry), (E) collagen and TRAP-induced aggregation (whole blood impedance aggregometry), and (F) thrombus formation was counteracted by IP receptor antagonist (RO-1138452 10 µM) given as a pretreatment before CXCR7 agonist for 15 minutes at room temperature. Data are mean ± SEM from 5 experiments with healthy donors. *P < .05, **P < .01, ***P < .001, ****P < .0001 with ANOVA followed by Sidak’s multiple comparison test. (Gi) Washed platelets from CAD patients (Table 2; n = 12) were treated with vehicle control (1% DMSO) or CXCR7 agonist (100 μg/mL) for 30 minutes at room temperature and processed for targeted lipidomics analysis for 12-HETrE. CXCR7 agonist treatment to CAD patient platelets ex vivo significantly increased 12-HETrE levels. Data are mean ± SEM. The significance levels for the FDR corrected P values are given using nonparametric paired Wilcoxon signed rank test with statistical significance *P < .05. (Gii-Giii) Inhibitory effect of CXCR7 agonist (100 µg/mL) observed in CAD patients on (Gii) platelet CD62P surface expression, PAC-1 binding (n = 5), and (Giii) TRAP induced aggregation (n = 6) was counteracted in presence of IP receptor antagonist (RO-1138452 10 µM) given as a pretreatment before CXCR7 agonist for 15 minutes at room temperature. In panels Gii through Giii, data are mean ± SEM; ####P < .0001 vs resting; §§§§P < .0001 vs CRP, *P < .02, ***P < .001; &&P < .002, &P < .02 with ANOVA followed by Sidak’s multiple comparison test. DMSO, dimethyl sulfoxide; Supernt., supernatant.
CXCR7 agonist–induced generation of antiplatelet lipid 12-HETrE and platelet inhibitory mediator cAMP. Washed platelets (200 × 106/sample) from n = 5 healthy donors were treated with CXCR7 agonist (100 µg/mL) for 0, 5, 15, and 30 minutes at room temperature. Thereafter platelet pellets were collected by centrifugation and used for detection of cAMP levels by LC-ESI-MS/MS analysis. (A) Gradual elevation in intraplatelet cAMP in response to CXCR7 agonist (100 µg/mL); *P < .05, **P < .01 vs 0 minute by Wilcoxon matched-pairs signed rank test, n = 5 donors. Washed human platelets (200 × 106/sample) from n = 5 healthy donors were treated with CXCR7 agonist (100 µg/mL) for 0, 5, 15, 30, and 60 minutes at room temperature. Intraplatelet levels of (Bi) linoleic acid (right y-axis), AA, and DGLA (left y-axis); (Bii) DGLA, 12-HETrE, and TxA(B)2; (Biii) linoleic acid (left y-axis) and 13-HODE (right y-axis); (Biv) EPA (left y-axis) and 12-HEPE (right y-axis); and (Bv) 12-HETrE, 12-HEPE, and 13-HODE as compared with TxA(B)2. #P < .05 and ##P < .01 vs AA; *P < .05, **P < .01 vs 0 minute using Welch’s t test. (C) Washed platelets (300 × 106/sample) from n = 9 healthy donors were treated with CXCR7 agonist (100 µg/mL) or vehicle control (1% DMSO) for 30 and 60 minutes at room temperature. IP receptor antagonist (RO-1138452 10 µM) was given as a pretreatment before CXCR7 agonist for 15 minutes at room temperature. At the end of incubation period, the supernatants were separated from the platelet pellets by centrifugation, and both were used for lipid extraction and subsequent targeted lipidomics analysis for 12-HETrE. Data are mean ± SEM. The significance levels for the corrected (FDR) P values are given using nonparametric paired Wilcoxon signed rank test with statistical significance *P < .05, **P < .01. Experiments done with blood from healthy donors (n = 5) showing inhibitory effect of CXCR7 agonist (100 µg/mL) on CRP (5 µg/mL) and TRAP (25 µM)-induced platelet (Di) PAC-1 binding, (Dii) CD62P surface expression (flow cytometry), (E) collagen and TRAP-induced aggregation (whole blood impedance aggregometry), and (F) thrombus formation was counteracted by IP receptor antagonist (RO-1138452 10 µM) given as a pretreatment before CXCR7 agonist for 15 minutes at room temperature. Data are mean ± SEM from 5 experiments with healthy donors. *P < .05, **P < .01, ***P < .001, ****P < .0001 with ANOVA followed by Sidak’s multiple comparison test. (Gi) Washed platelets from CAD patients (Table 2; n = 12) were treated with vehicle control (1% DMSO) or CXCR7 agonist (100 μg/mL) for 30 minutes at room temperature and processed for targeted lipidomics analysis for 12-HETrE. CXCR7 agonist treatment to CAD patient platelets ex vivo significantly increased 12-HETrE levels. Data are mean ± SEM. The significance levels for the FDR corrected P values are given using nonparametric paired Wilcoxon signed rank test with statistical significance *P < .05. (Gii-Giii) Inhibitory effect of CXCR7 agonist (100 µg/mL) observed in CAD patients on (Gii) platelet CD62P surface expression, PAC-1 binding (n = 5), and (Giii) TRAP induced aggregation (n = 6) was counteracted in presence of IP receptor antagonist (RO-1138452 10 µM) given as a pretreatment before CXCR7 agonist for 15 minutes at room temperature. In panels Gii through Giii, data are mean ± SEM; ####P < .0001 vs resting; §§§§P < .0001 vs CRP, *P < .02, ***P < .001; &&P < .002, &P < .02 with ANOVA followed by Sidak’s multiple comparison test. DMSO, dimethyl sulfoxide; Supernt., supernatant.
The apparent paradox of cAMP elevation was clarified by exploring the platelet lipidome following ACKR3/CXCR7 ligation using a targeted approach for oxylipins.43 We observed a time-dependent increase in intraplatelet levels of linoleic acid, AA, the antiplatelet lipid dihomo-γ-linolenic acid (DGLA) (Figure 6Bi), and a subsequent rise in the levels of DGLA-derived 12-LOX metabolite 12-HETrE (Figure 6Bii). Levels of linoleic acid–derived CytP450 or COX-1 metabolite 13-hydroxyoctadecadienoic acid (13-HODE) (Figure 6Biii) and eicosapentanoic acid (EPA)-derived 12-LOX metabolite 12-HEPE (Figure 6Biv) with reported antiplatelet effects63 were also increased but that of AA-derived prothrombotic COX-1 metabolite TxA(B)2 remain unaltered (Figure 6Bv).
Platelet membranes contain more AA than DGLA,63 which may be metabolized into prothrombotic or antithrombotic lipids. To get a translational perspective, we checked the relative levels of thrombogenic AA and antiplatelet DGLA in platelets from CAD (n = 107) patients receiving APT and statin. Comparative levels of AA (mean ± SD, without APT: 513 ± 297 ng/mL, with APT: 661 ± 455 ng/mL) and DGLA (without APT: 61 ± 37 ng/mL, with APT: 76 ± 63 ng/mL) were observed in CAD patients despite APT (supplemental Figure 6Di), as well as among those undergoing statin therapy (supplemental Figure 6Dii). Neither DGLA (CCS: 70 ± 53 ng/mL, ACS: 59 ± 33 ng/mL) nor AA (CCS: 600 ± 384 ng/mL, ACS: 526 ± 335 ng/mL) levels altered with disease severity (supplemental Figure 6Ei), irrespective of APT administration (supplemental Figure 6Eii) (DGLA: CCS, 75 ± 57 ng/mL, ACS, 59 ± 43 ng/mL; AA: CCS, 647 ± 406 ng/mL, ACS, 576 ± 467 ng/mL) (supplemental Excel File 1).
The antithrombotic actions of 12-HETrE released from platelets are mediated through Gαs-coupled prostacyclin receptor (IP).64 We observed a time-dependent increase in generation and release of 12-HETrE from CXCR7 agonist–treated platelets (Figure 6C). Moreover, presence of IP antagonist prevented 12-HETrE from binding back to the IP receptor, which consequently increased levels of 12-HETrE in the platelet supernatant. This clarified the unexpected rise in cAMP as mediated by 12-HETrE acting through Gαs-coupled IP receptor in coordination with ACKR3/CXCR7 ligation. Therefore, IP antagonist decreased the inhibitory effects of CXCR7 agonist on-platelet activation (Figure 6Di-Dii), aggregation (Figure 6E), and thrombus formation (Figure 6F) without affecting basal platelet response to activating stimuli or the platelet lipidome (supplemental Figure 7Bi-Bii). Inhibitors of adenylyl cyclase (SQ22536) (supplemental Figure 8A) and cAMP-dependent kinases (KT5720) (supplemental Figure 8B) also counteracted the inhibitory actions of CXCR7 agonist on-platelet activation, aggregation, and thrombus formation. As in platelets from healthy subjects (Figure 6B-C), CXCR7 agonist treatment ex vivo induced 12-HETrE generation in platelets from CAD patients (Figure 6Gi) whereas IP antagonist counteracted the inhibitory actions of CXCR7 agonist on-platelet activation (Figure 6Gii) and aggregation (Figure 6Giii). Therefore, an alternate antiplatelet strategy through ACKR3/CXCR7 ligation counteracting generation of thrombogenic COX-1 (TxA2, HHT) or 12-LOX (12-HETE) metabolites in favor of antiplatelet lipids (12-HETrE, 13-HODE, 12-HEPE) might be considered advantageous.
CXCR7 agonist regulates HIT-associated thrombotic and thromboinflammatory response
Thrombo-inflammation exaggerates micro/macrothrombotic consequences.5 Since ACKR3/CXCR7 ligation counteracted inflammatory release and platelet-leukocyte interactions, we verified its regulatory potential against thrombotic and thromboinflammatory challenges inflicted by HIT+ sera/IgG. HIT is a drug-induced thromboinflammatory adversity that may cause venous/arterial thrombosis. Upon heparin administration in emergency during a thromboischemic event (MI), susceptible individuals develop anti–PF4-heparin antibodies that activate platelets through FcγRIIA, necessitating heparin replacements.
Sera from HIT+ patients significantly enhanced platelet (Figure 7A-D) and neutrophil (Figure 7F) activation. CXCR7 agonist reduced CD62P expression (Figure 7A), αIIbβ3-integrin activation (Figure 7B), platelet response in HIPA test (Figure 7C), and thrombus formation (Figure 7D) triggered by HIT+ sera ex vivo. 12-LOX plays a vital role in mediating prothrombotic response triggered through FcγRIIA.58,65 Moreover, both TxA266 and 12-HETE67 have prominent proinflammatory attributes. Therefore, 12-LOX is a potential antiplatelet drug target,63 like COX-1. CXCR7 agonist significantly reduced HIT sera-induced generation of thromboinflammatory COX-1–derived TxA2 and platelet 12-LOX-derived 12-HETE (Figure 7E), which could have contributed to its antithrombotic effects (Figure 7A-D) in identical experimental settings ex vivo.
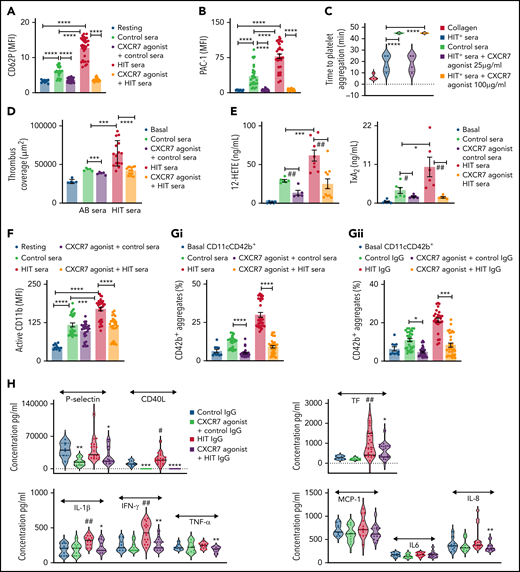
CXCR7 agonist counteracts thrombotic and thromboinflammatory response elicited by HIT sera/IgG. Whole blood assays with heparinized (0.2 IU/mL) blood from healthy donors (n = 5) was carried out in presence of control sera (n = 3) and HIT+ patient sera (n = 3) incubated at 1:10 vol/vol dilution for 1 hour at room temperature. CXCR7 agonist (100 µg/mL) or vehicle control (1% DMSO) was given as a pretreatment for 30 minutes at room temperature. HIT+ patient sera induced (A) platelet CD62P surface expression and (B) PAC-1 binding denoting αIIbβ3-integrin activation ex vivo. (C) HIPA assay performed with isolated washed platelets from 3 healthy donors in presence/absence of CXCR7 agonist (25 and 100 µg/mL) or vehicle control shows an inhibitory effect of CXCR7 agonist. (D) Ex vivo thrombus formation was performed in heparinized (0.2 IU/mL) blood from healthy donors (n = 5) over collagen-coated surface in presence of control sera (n = 3) and HIT+ patient sera (n = 3) in presence/absence of CXCR7 agonist (100 µg/mL) or vehicle control. In panels A through D, *P < .05, ***P < .001, ****P < .0001 using ANOVA followed by Sidak’s multiple comparison test. (E) Targeted lipidomics analysis for oxylipins showed that ex vivo treatment with control sera (n = 3), HIT+ patient sera (n = 3), HIT+ patient sera (n = 3) for 1 hour at room temperature induced generation of COX-1–derived TxA2 and platelet 12-LOX–derived 12-HETE, which was significantly counteracted in the presence of CXCR7 agonist (100 µg/mL) given as a pretreatment of 30 minutes at room temperature. *P < .05, #P < .05, ##P < .01, ***P < .001 with Wilcoxon signed rank test. (F) Neutrophil CD11b (anti-human active CD11b-FITC) activation ex vivo in heparinized (0.2 IU/mL) blood from healthy donors (n = 5) in response to control sera (n = 3) or sera from HIT+ patients (n = 3) (at 1:10 vol/vol dilution) in presence/absence of CXCR7 agonist (100 µg/mL) or vehicle control. Diagram showing formation of CD42b+ (anti-human CD42b-FITC)-CD11c+ (anti-human CD11c-APC) platelet-neutrophil aggregates ex vivo in heparinized (0.2 IU/mL) blood from healthy donors (n = 5 donors) treated with (Gi) sera or (Gii) corresponding isolated IgG fractions from control healthy subjects and HIT+ patients (n = 3 each) at 1:10 vol/vol dilution for 1 hour at room temperature in presence/absence of CXCR7 agonist (100 µg/mL). In panels F through G, *P < .05, ***P < .001, ****P < .0001 using ANOVA followed by Sidak’s multiple comparison test. Washed human platelets (200 × 106/sample) from n = 5 donors were pretreated with CXCR7 agonist (100 µg/mL) or vehicle control for 30 minutes at room temperature and then with IgG fractions from control or HIT+ patient sera (n = 3 each) at 1:10 vol/vol for 1 hour at room temperature. Activated platelet supernatants were collected and analyzed by cytometric bead array (Legendplex 13-plex human inflammation panel, 10-plex human thrombosis panel). (H) Violin plots (solid line denotes median) showing levels of proinflammatory mediators and chemoattractants in platelet releasate. Data represent mean ± SEM; #P < .05, ##P < .01 vs control IgG; *P < .05, **P < .01, ***P < .001, ****P < .0001 vs HIT+ IgG using Mann-Whitney U test for each inflammatory mediator.
CXCR7 agonist counteracts thrombotic and thromboinflammatory response elicited by HIT sera/IgG. Whole blood assays with heparinized (0.2 IU/mL) blood from healthy donors (n = 5) was carried out in presence of control sera (n = 3) and HIT+ patient sera (n = 3) incubated at 1:10 vol/vol dilution for 1 hour at room temperature. CXCR7 agonist (100 µg/mL) or vehicle control (1% DMSO) was given as a pretreatment for 30 minutes at room temperature. HIT+ patient sera induced (A) platelet CD62P surface expression and (B) PAC-1 binding denoting αIIbβ3-integrin activation ex vivo. (C) HIPA assay performed with isolated washed platelets from 3 healthy donors in presence/absence of CXCR7 agonist (25 and 100 µg/mL) or vehicle control shows an inhibitory effect of CXCR7 agonist. (D) Ex vivo thrombus formation was performed in heparinized (0.2 IU/mL) blood from healthy donors (n = 5) over collagen-coated surface in presence of control sera (n = 3) and HIT+ patient sera (n = 3) in presence/absence of CXCR7 agonist (100 µg/mL) or vehicle control. In panels A through D, *P < .05, ***P < .001, ****P < .0001 using ANOVA followed by Sidak’s multiple comparison test. (E) Targeted lipidomics analysis for oxylipins showed that ex vivo treatment with control sera (n = 3), HIT+ patient sera (n = 3), HIT+ patient sera (n = 3) for 1 hour at room temperature induced generation of COX-1–derived TxA2 and platelet 12-LOX–derived 12-HETE, which was significantly counteracted in the presence of CXCR7 agonist (100 µg/mL) given as a pretreatment of 30 minutes at room temperature. *P < .05, #P < .05, ##P < .01, ***P < .001 with Wilcoxon signed rank test. (F) Neutrophil CD11b (anti-human active CD11b-FITC) activation ex vivo in heparinized (0.2 IU/mL) blood from healthy donors (n = 5) in response to control sera (n = 3) or sera from HIT+ patients (n = 3) (at 1:10 vol/vol dilution) in presence/absence of CXCR7 agonist (100 µg/mL) or vehicle control. Diagram showing formation of CD42b+ (anti-human CD42b-FITC)-CD11c+ (anti-human CD11c-APC) platelet-neutrophil aggregates ex vivo in heparinized (0.2 IU/mL) blood from healthy donors (n = 5 donors) treated with (Gi) sera or (Gii) corresponding isolated IgG fractions from control healthy subjects and HIT+ patients (n = 3 each) at 1:10 vol/vol dilution for 1 hour at room temperature in presence/absence of CXCR7 agonist (100 µg/mL). In panels F through G, *P < .05, ***P < .001, ****P < .0001 using ANOVA followed by Sidak’s multiple comparison test. Washed human platelets (200 × 106/sample) from n = 5 donors were pretreated with CXCR7 agonist (100 µg/mL) or vehicle control for 30 minutes at room temperature and then with IgG fractions from control or HIT+ patient sera (n = 3 each) at 1:10 vol/vol for 1 hour at room temperature. Activated platelet supernatants were collected and analyzed by cytometric bead array (Legendplex 13-plex human inflammation panel, 10-plex human thrombosis panel). (H) Violin plots (solid line denotes median) showing levels of proinflammatory mediators and chemoattractants in platelet releasate. Data represent mean ± SEM; #P < .05, ##P < .01 vs control IgG; *P < .05, **P < .01, ***P < .001, ****P < .0001 vs HIT+ IgG using Mann-Whitney U test for each inflammatory mediator.
Neutrophils aggravate HIT-associated thrombotic complications.8 CXCR7 agonist significantly decreased neutrophil CD11b activation (Figure 7F) triggered by HIT+ sera. Combined with its effect on platelet CD62P exposure and αIIbβ3-integrin activation that engage in interaction with P-selectin glycoprotein ligand (PSGL-1) and Mac-1 complex (CD11b/CD18), CXCR7 agonist significantly reduced platelet-neutrophil aggregate formation triggered by HIT+ sera and IgG fractions (Figure 7Gi-Gii; supplemental Figure 9); moreover, it counteracted release of thromboinflammatory mediators (eg, IL-1β, IFN-γ, sCD40L, TNF-α, sP-selectin, neutrophil chemoattractant IL-8, and thrombogenic TF) from HIT+ IgG-treated platelets (Figure 7H). Therefore, therapeutics targeting ACKR3/CXCR7 may be useful to cope with HIT-associated thromboinflammation, as well as in acute inflammatory pathologies that trigger platelet FcγRIIA, as in vaccine-induced immune thrombotic thrombocytopenia68 that closely resembles HIT or antibodies against SARS-CoV2.3,6
Discussion
Capitalizing on our extensive work exploring platelet CXCL12/SDF1α-CXCR4- ACKR3/CXCR7 axis in experimental26,27 and clinical studies,16,17,29,48 novel translational findings of the current investigation are (1) Platelet ACKR3/CXCR7 surface expression is enhanced post-MI, which could be therapeutically targeted by a CXCR7 agonist to reduce thrombotic response post-MI in vivo (murine model) and ex vivo (ACS patients). (2) CXCR7 agonist checked arterial thrombosis in vivo without compromising basal hemostasis. (3) CXCR7 agonist also regulated thromboinflammatory platelet response triggered by HIT+ sera/IgG ex vivo and in peripheral circulation of mice post-MI and arterial injury in vivo. (4) ACKR3/CXCR7 ligation modulated the platelet lipidome to limit metabolism and release of thrombotic and atherogenic lipid mediators while favoring the generation of antiplatelet lipids in healthy subjects and CAD patients ex vivo. (5) 12-HETrE–mediated engagement of Gαs-coupled IP receptor following ACKR3/CXCR7 ligation triggered the platelet inhibitory AC-cAMP–protein kinase A cascade to regulate thromboinflammatory functions.
ACKR3/CXCR7 primarily as an atypical/noncanonical GPCR shows cell- and ligand-specific functional response.32 Recently, the cardiovascular significance of ACKR3/CXCR7 has become evident in the hematopoietic-vascular niche,33 platelets,16,17,26,27,48 atherosclerosis,34 and CAD.16,17,29,48 Its therapeutic potential has been shown in murine models of myocardial infarction,18,19,21 atherosclerosis,34 and in impeding pulmonary fibrosis33 by employing pharmacological CXCR7 agonists32 and lineage-specific cxcr7-deficient mice.19,34 Systemic administration of CXCR7 agonist in the current investigation may have exerted significant impact on both the target organ (myocardium), as previously described,19,21 and cells in peripheral circulation, including platelets. The currently observed antithrombotic effect adds to its therapeutic value. Differences in relative platelet CXCR4 and ACKR3/CXCR7 availability between murine MI models, stable CAD, and ACS patients put aside, platelet ACKR3/CXCR7 surface expression is low under physiological conditions,26,27 as are levels of its physiological ligands (CXCL12/SDF1-α, MIF). However, a significant increment in platelet ACKR3/CXCR7 is observed post-MI in mice and ACS patients. ACKR3/CXCR7 is not directly involved in executing thrombotic-hemostatic platelet functions. Therefore, deletion of cxcr7 from murine platelets may not exhibit a drastic platelet-centered phenotype under physiological conditions. Attempting to close the translational gap between experimental and clinical research, we validated our observations in CAD patients exhibiting increased platelet ACKR3/CXCR7 surface expression16,17,48 with experimental evidence from human system ex vivo/in vitro and murine models in vivo. Antithrombotic effects of CXCR7 agonist ascertained in whole blood from ACS patients, closely resembling physiological conditions ex vivo, corroborated with those detected post-MI and arterial injury in CXCR7 agonist administered mice in vivo, offering the possibility of preclinical validation. Enhanced platelet ACKR3/CXCR7 availability post-MI may regulate platelet response and reduce thromboinflammatory platelet-leukocyte interactions in circulation in presence of an adequate ligand. Considering increased formation of platelet-monocyte aggregates in ACS patients, attributed to elevated sP-selectin, IL-6 levels, and its association with enhanced risk of ACS,7 CXCR7 agonist offered regulation on platelet CD62P, plasma IL-6, and platelet-leukocyte aggregate formation post-MI seem favorable.
Cxcr7-deficient hyperlipidemic mice show increased neointima formation and macrophage accumulation at carotid lesions, whereas administration of CXCR7 agonist (CCX771) ablates atheroprogression34 and reduces plasma cholesterol by prompting uptake of very-low-density lipoproteins in the adipose tissue.34 Currently, we evidenced an unprecedented influence of ACKR3/CXCR7 ligation on the platelet lipidome in reducing the generation of prothrombotic LPCs, LPIs, and DGs, levels of which are elevated in platelets from CAD patients,29 notwithstanding antiplatelet or statin therapy. In thrombin-activated platelets from healthy subjects and CAD patients alike, CXCR7 agonist also reduced the generation and release of AA, HHT, TxA2, and 12-HETE. Such a unique influence of ACKR3/CXCR7 may check atherothrombosis by regulating generation of prothrombotic lipids and atherogenic LPCs carried by platelets and platelet-derived microvesicles.60 However, this needs to be experimentally validated in atherosclerosis-prone murine models. Apparently, CXCR7 agonist–mediated effects have an unexpected advantage over ASA, which inhibits COX-1 metabolism downstream of phospholipases, whereas levels of LPIs, LPCs, and DGs remain elevated in CAD patients,29 and thrombogenic 12-HETE and HETE phospholipids can be formed by 12-LOX.59 Moreover, CXCR7 agonist potentiated the generation of antiplatelet lipids (DGLA, 12-HETrE,64 13-HODE, 12-HEPE),63 interestingly limiting 12-LOX–metabolized prothrombotic, procoagulatory AA-derived 12-HETE but promoting DGLA- and EPA-derived antiplatelet 12-LOX products (ie, 12-HETrE and 12-HEPE, respectively). Although mechanistic reason behind this preference remains to be deciphered, a deliberate shift favoring antiplatelet lipids seems promising. Attributed to 12-HETrE, ACKR3/CXCR7 ligation coordinated with Gαs-coupled IP receptor to elevate cAMP levels and regulate thrombotic functions, an effect contrary to the prothrombotic actions of platelet CXCR4.29-31 Gαi-coupled CXCR4 triggers a canonical signaling cascade following CXCL12/SDF-1α binding that decreases cAMP levels30,31 while inducing intracellular calcium mobilization, PI3K-Akt activation, and release of ATP and TxA2, which is contrary to the influence of ACKR3/CXCR7 on platelet activatory signaling cascade (visual/graphical abstract).
Acute or chronic thromboinflammatory processes may cause cardiovascular complications.1,2,5 Impact of antiplatelet therapies on inflammatory mediators like hsCRP, IL-6, and TNF-α has been deduced in clinical studies.14 Currently, CXCR7 agonist decreased thromboinflammatory release from CRP (GPVI)- and thrombin (PAR)-activated platelets and those triggered by HIT+ IgG (FcγRIIA). ACKR3/CXCR7 ligation also counteracted HIT+ sera/IgG-induced platelet and neutrophil activation, platelet-neutrophil interactions, and generation of proinflammatory oxylipins TxA266 and 12-HETE.67 Apart from its antiplatelet effects, CXCR7 agonist may also influence inflammatory functions of ACKR3/CXCR7-expressing immune cells, epithelium, and endothelium,69 much like the effect of antiplatelet ASA on COX-expressing cells. Considering the inhibitory impact of CXCR7 agonist (VUF11207) on thrombotic and thromboinflammatory functions, this opens a new vista of investigation in validating the therapeutic potential of ACKR3/CXCR7 in acute/chronic inflammatory pathologies that trigger thrombotic complications.5
Presently, we have substantiated our previously recognized16,17,23,27 translational implications of ACKR3/CXCR7 in platelet pathophysiology. In CAD patients, increased platelet CXCR4 and ACKR3/CXCR7 expression is associated with improved prognosis.16,17 CXCR4 is relatively more abundant on platelets under physiological conditions,26 whereas ACKR3/CXCR7 might be transiently induced post-MI16 by circulatory or platelet-associated CXCL12/SDF1α in ACS patients15,24 as observed in experimental studies.26 ACKR3/CXCR7 may offer a novel therapeutic paradigm to fine-tune thrombotic and thromboinflammatory platelet response without affecting physiological/basal hemostasis or antagonizing the function and surface availability of platelet receptors. There is no evidence that ACKR3/CXCR7 interacts with these receptors or their respective ligands. Therefore, pharmacological targeting of ACKR3/CXCR7 to overcome platelet-driven thromboinflammation may diverge from the direct inhibitory effects of antiplatelet therapeutics that act as receptor antagonists (P2Y12, GPIIbIIIa, PAR-1 antagonists). Translational implications of exploiting ACKR3/CXCR7 may be realized by (a) assessing the therapeutic “time window” of increased ACKR3/CXCR7 availability as a potential drug target on circulating platelets post-MI in longitudinal clinical studies with ACS patients and (b) by extending its therapeutic validation in HIT and other thromboinflammatory pathologies.
Acknowledgments
This work was supported by funding from the German Research Foundation (DFG) project number 374031971-TRR240, Deutsche Stiftung für Herzforschung (DSHF-F/22/17) to M. Chatterjee, and DFG project number BO3786/3-1 to O.B.
Authorship
Contribution: M. Cebo, K.D., and X.F. performed lipidomics analysis; M.C.M. performed intravital microscopy (IVM); F.E. performed thromboelastography and Legendplex analysis; J.R. and H.v.E. performed SICM; N.F. analyzed cAMP levels; J.S. analyzed the MI/RI model; A.W. analyzed tail bleeding time; L.P. characterized HIT patients; D.R. analyzed clinical data; T.E.S. supervised SICM; M.L. supervised lipidomics analysis with M. Chatterjee; O.B. (IVM), T.G. (CAD cohort), T.B. (HIT), and M.G. (MI/RI model) collaborated in the project; and M. Chatterjee was involved in the project design, supervision, experimentation, data analysis, and manuscript writing.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Madhumita Chatterjee, Department of Cardiology and Angiology, University Hospital Tübingen, Otfried-Müller-Straße 10, 72076, Tübingen, Germany; e-mail: madhumita.chatterjee@med.uni-tuebingen.de.
For original data, please contact madhumita.chatterjee@med.uni-tuebingen.de.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal