

Functional decline upon repeated stimulation, or exhaustion, is the bugbear of T-cell–based immunotherapies. Although combining chimeric antigen receptor (CAR) T cells with immune checkpoint inhibitors is an attractive concept, it is not clear that this approach can rescue already dysfunctional T cells because deep exhaustion is epigenetically encoded1 and poorly reversible. In this issue of Blood, Yoshikawa et al2 ablate a transcription factor that is known to repress T-cell memory formation, leading to enhanced memory phenotype and cytokine polyfunctionality of tumor-specific T cells.
PR domain zinc finger protein-1 (PRDM1) encodes Blimp-1, a known repressor of T-cell memory formation. Adoptive immunotherapy using CAR-engineered T cells, T-cell receptor–engineered T cells, and tumor-infiltrating lymphocytes (TILs) is efficacious when T cells have prolonged persistence,3 whereas terminal differentiation of T cells and subsequent loss of memory phenotype hinder robust antitumor response.4 In this article, the authors demonstrate a novel approach of epigenetically reprogramming T-cell memory state in antitumor T cells by genetically ablating PRDM1 (Blimp-1), a known repressor of T-cell memory.5
The transcription factor Blimp-1 is known to drive differentiation, exhaustion, and suppression of memory formation in T cells by downregulating expression of memory-related genes.5 The authors deleted PRDM1 in CAR19 T cells using CRISPR-Cas9 and induced progressive dysfunction by repeated stimulation with CD19-expressing target cells. PRDM1 deletion in CAR19 T cells resulted in increased central memory T-cell (TCM) and memory stem T-cell (TSCM) subsets upon antigen restimulation compared with the control. However, PRDM1 deletion did not affect the proliferation of T cells. PRDM1-deficient T cells demonstrated secretion of multiple cytokines like interleukin-2, interferon-γ, and tumor necrosis factor-α. The authors then demonstrated improved persistence, a superior antitumor effect, and increased TCM and TSCM in PRDM1-deficient CAR19 T cells in vivo. PRDM1 deficiency resulted in elevated expression of memory-associated transcription factors and surface markers, and downregulation of effector differentiation genes correlating with increased chromatin accessibility of key genes involved in memory formation (see figure). Thus, using a gene-editing strategy, the authors succeeded in epigenetically engineering the T-cell memory state.
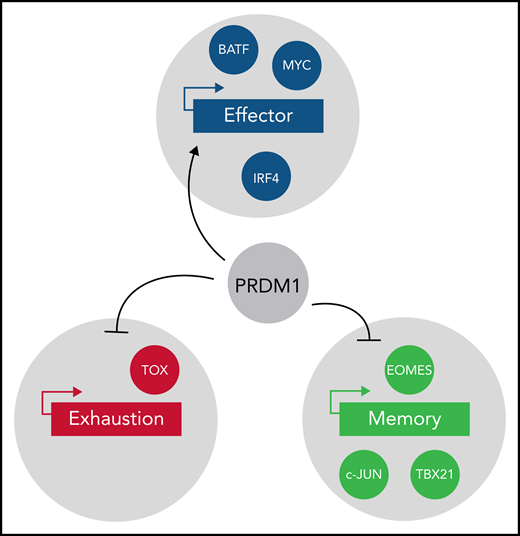
Several transcription factors are known to modulate effector function (blue), exhaustion (red), and memory phenotype (light green) in T cells. Yoshikawa et al clearly establish PRDM1 as a repressor of memory function in antitumor T cells. PRDM1 deletion resulted in upregulation of TOX and EOMES in T cells. EOMES upregulation in PRDM1-deficient T cells partially explains the promotion of memory phenotype. However, upregulation of TOX in PRDM1-deficient T cells further raises interesting questions regarding epigenetic programs linking memory and exhaustion.
Several transcription factors are known to modulate effector function (blue), exhaustion (red), and memory phenotype (light green) in T cells. Yoshikawa et al clearly establish PRDM1 as a repressor of memory function in antitumor T cells. PRDM1 deletion resulted in upregulation of TOX and EOMES in T cells. EOMES upregulation in PRDM1-deficient T cells partially explains the promotion of memory phenotype. However, upregulation of TOX in PRDM1-deficient T cells further raises interesting questions regarding epigenetic programs linking memory and exhaustion.
Others have recently overexpressed AP-1 family transcription factors, such as BATF6 or c-JUN,7 to prevent the development of T-cell exhaustion. However, Yoshikawa and colleagues went further here by (partially) reprogramming the memory phenotype in already differentiated T cells. They did this by deleting PRDM1 in prestimulated CAR19 T cells or TILs extracted from patients with gynecologic or lung cancers. PRDM1 deficiency in prestimulated CAR19 T cells and TILs partially restored expression of memory markers. This reveals the importance of the timing of targeting an epigenetic modulator and suggests that additional factors might be involved in maintaining the repressed chromatin state of certain memory genes. Notably, elimination of PRDM1 was effective despite the fact that it resulted in the upregulation of the exhaustion-driving thymocyte-selection associated high-mobility group box (TOX)8 transcription factor and regardless of the fact that PRDM1-deficient T cells showed higher expression of typical surface markers of exhaustion.
The translational potential of this approach is underscored by a recently published experiment of nature, wherein inadvertent biallelic disruption of an epigenetic modulator, TET2, in a patient receiving CAR T cells for the treatment of chronic lymphocytic leukemia resulted in massive expansion of a single T cell and durable remission.9 However, because in the current article the reprogramming of prestimulated T cells into memory T cells was partial, future work could focus on identifying barriers to an even more profound effect.
Manipulation of inhibitory immune receptors and transcription factors with roles in T-cell exhaustion are some of the proposed strategies for generating efficacious CAR T cells. Because the T-cell exhaustion epigenetic program is intricately linked with memory formation, such strategies will likely come at the cost of impaired long-term memory. The work presented here suggests that engineering a robust memory phenotype in CAR T cells is a more pragmatic and efficacious strategy rather than focusing on preventing exhaustion per se.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal