


Abstract
Polymorphonuclear neutrophils (PMNs) figure prominently in host defense against infection and in noninfectious inflammation. Mobilized early in an inflammatory response, PMNs mediate immediate cellular defense against microbes and orchestrate events that culminate in cessation of inflammation and restoration of homeostasis. Failure to terminate the inflammatory response and its causes can fuel exuberant inflammation characteristic of many human diseases, including cystic fibrosis (CF), an autosomal recessive genetic disease caused by mutations in the CF transmembrane conductance regulator. CF affects multiple end organs, with persistent bacterial infection and chronic neutrophilic inflammation in airways predominating the clinical picture. To match the diverse microbial challenges that they may encounter, PMNs possess a variety of antimicrobial systems to slow or kill invading microorganisms confined in their phagosomes. Prominent among PMN defense systems is their ability to generate hypochlorous acid, a potent microbicide, by reacting oxidants generated by the NADPH oxidase with myeloperoxidase (MPO) released from azurophilic granules in the presence of chloride (Cl−). Products of the MPO-H2O2-Cl system oxidize susceptible biomolecules and support robust antimicrobial action against many, but not all, potential human pathogens. Underscoring that the MPO-H2O2-Cl system is integral to optimal host defense and proper regulation of inflammation, individuals with defects in any component of this system, as seen in chronic granulomatous disease or MPO deficiency, incur increased rates or severity of infection and signs of dysregulated inflammatory responses. We focus attention in this review on the molecular basis for and the clinical consequences of defects in the MPO-H2O2-Cl system because of the compromised Cl transport seen in CF. We will discuss first how the MPO-H2O2-Cl system in healthy PMNs participates in host defense and resolution of inflammation and then review how a defective MPO-H2O2-Cl system contributes to the increased susceptibility to infection and dysregulated inflammation associated with the clinical manifestations of CF.
Introduction
The human immune system serves to protect host integrity from compromise by infection, trauma, attack on self, and other external forces. Secreted soluble agents and cells, both circulating and tissue-based, collaborate to recognize danger, recruit responsive elements, rebuff threats, and restore homeostasis. Polymorphonuclear neutrophils (PMNs) play a central role in host defense against microbes, contributing both to resolution of infection and to restoration of the baseline.1-3 PMNs, the most abundant leukocytes in circulation and the first responder cells to arrive at sites of infection or injury, eliminate their targets via phagocytosis and confine them to phagosomes, where microbes are exposed to an array of bioactive molecules released into and generated within the phagosome. Synergistic interactions among these PMN-derived agents compromise the integrity and viability of ingested microbes. Concomitant with the initiation of antimicrobial responses, the process of phagocytosis prompts transcription of genes that accelerate apoptosis of PMNs and culminate in termination of the inflammatory response.4
Among the many mediators of PMN responses to microbes are the products of the reactions of the azurophilic granule protein myeloperoxidase (MPO), H2O2 generated by the NADPH oxidase, and chloride anion shuttled from cytoplasm into the phagosome.1 The occurrence of relative immune deficiency in individuals with inherited or acquired defects in any constituent of the system underscores the clinical significance of the MPO-H2O2-Cl system for host defense. In addition, the failure to restore homeostasis after an inflammatory response leads to exaggerated clinical manifestations or the genesis of autoinflammatory diseases.
The MPO-H2O2-Cl system is particularly germane to the subject of innate immunity in cystic fibrosis (CF).5 Because deficiencies in MPO (ie, MPO deficiency) and in the NADPH oxidase (ie, chronic granulomatous disease) have been extensively reviewed,6-8 we will focus attention in this review on the molecular basis for and the clinical consequences of defects in the MPO-H2O2-Cl system caused by the deficiency of Cl transport seen in CF. We will discuss first how the MPO-H2O2-Cl system in healthy PMNs participates in host defense and resolution of inflammation and then review how a defective MPO- H2O2-Cl system contributes to the pathogenesis of CF.
PMNs in normal human host defense against infection
A normal adult human generates 1 to 2 × 1011 PMNs daily, with a small fraction entering the circulation, and the majority reserved in the bone marrow for emergency needs.9 PMNs are highly specialized in the capture, engulfment, and killing of a wide variety of microorganisms. Attesting to the importance of PMNs in human health, patients with inadequate quantity or compromised quality of circulating PMNs, such as in bone marrow failure,10 radio- or chemotherapy,11 or inherited disorders of PMNs,12,13 exhibit increased susceptibility to infection. To fulfill their host defense function and to cope with the diverse microbes they may encounter, PMNs generate a myriad of bioactive agents that are microbicidal and can be categorized into one of two groups based on their origin. One group includes proteins synthesized in myeloid precursors during granulopoiesis and stored in granules.14 These granule proteins include serine proteases (azurocidin, proteinase-3, cathepsin G, and elastase), MPO, defensins, the bactericidal/permeability-increasing protein, cathelicidins, lactoferrin, and gelatinase. The other group of bioactive agents includes reactive oxygen species produced de novo at the time of PMN activation by the multicomponent NADPH oxidase.15 To understand the contribution of PMNs to normal host defense against infection, it is critical to recognize that none of these agents acts alone, but rather, effective microbicidal activity of human PMN depends on the collective action of proteins delivered by fusion of PMN granules with nascent phagosomes and oxidants generated by the NAPDH oxidase. A successful response to infection culminates in elimination of the invading microbe and restoration of homeostasis, as inflammatory responses that persist after the elimination of microbes will exact damage on the host.
Phagocyte NADPH oxidase as a source of oxidants in PMNs
The phagocyte NADPH oxidase is a multicomponent protein complex composed of a heterodimeric transmembrane protein, NOX2-p22phox (aka gp91phox-p22phox), and several cytosolic proteins that translocate to dock at the abluminal side of the phagosome.15 The assembled complex operates as an electron transferase that transports electrons from cytosolic NADPH into the phagosomal lumen, where molecular oxygen undergoes single electron reduction to generate superoxide anion, the most proximal product of the NADPH oxidase.16 Phagocytosing PMNs at their maximal capacity consume ∼10 nmoles oxygen per minute per million cells.17,18 Uncompensated, a unidirectional electron transfer would rapidly generate a transmembrane potential, which eventually would terminate electron transport and oxidant generation.19 However, the voltage-gated proton channel Hv1 is activated when the membrane potential reaches a certain threshold, thus allowing protons to passively flow from the cytoplasm into the phagosomal lumen and thereby neutralize the potential.19-24 Superoxide anion, the proximal product of the NADPH oxidase, is short-lived and consumed in the generation of other oxidants, including H2O2, a moderate oxidant, through a spontaneous or MPO-catalyzed dismutation.25
MPO uses H2O2 to oxidize chloride to produce chlorine bleach (HOCl)
The azurophilic granules of human PMNs possess significant amounts of MPO, which accounts for 2% to 5% of the dry weight of human PMNs.26 Monocytes have approximately one-fifth of the MPO present in PMNs, but monocyte-derived macrophages express almost none. In general, all mammalian heme peroxidases, namely MPO, eosinophil peroxidase, lactoperoxidase, and thyroid peroxidase, can oxidize halides (and the pseudohalide SCN−) depending on their redox potentials, with SCN− > I− > Br− > Cl− in descending order of ease.27 Extensive investigation by Klebanoff28-30 established that peroxidase-mediated halogenation of microbes can be microbicidal and that MPO-dependent attack of microbial targets contributes to neutrophil microbicidal function. The recovery of chlorinated microbes from PMN phagosomes demonstrates that MPO-driven chemistry could promote killing of ingested bacteria. HOCl is rapidly consumed in reactions with proteins in phagosomes, both from granules and from microbes, and many of the downstream products, such as chloramines, are themselves microbicidal and some long-lived.31,32
MPO is unique among all mammalian peroxidases because of its high reduction potential. At pH 7.0, the standard redox potential for generation of Compound I, the oxidizing product of the reaction of MPO with H2O2, is 1.16 V.33,34 Thus, MPO is unique among the mammalian heme peroxidases in its ability at physiologic pH to catalyze the oxidation of chloride to HOCl, the active component in common laundry bleach.30 As the nascent phagosomes arises during phagocytosis, superoxide anion generated by the NAPDH oxidase, MPO delivered by fusion of azurophilic granules with the nascent phagosome, and Cl− transport from the cytosol support production of the potent microbicide HOCl (Figure 1). Native MPO is in the ferric state (Fe+3) and supports two reactions essential for the production of HOCl within the PMN phagosome. MPO dismutates superoxide anion to H2O2, which in turn reacts with native MPO to generate Compound I,25 a potent but transient oxidant that mediates the 2-electron oxidation of Cl− to generate HOCl.17 Specific structural features around the heme pocket of MPO enable the enzyme to execute this highly specialized chemistry.34
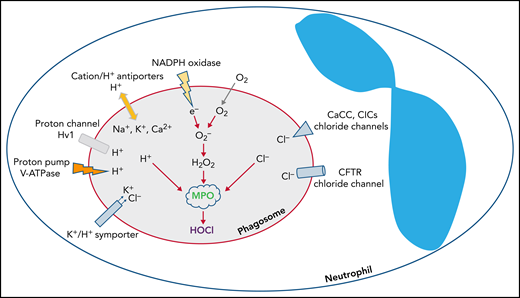
Chloride is the predominant anion that regulates phagosomal oxidant production in PMN phagosomes. NADPH oxidase transfers electrons into the phagosomal lumen, thereby creating a transmembrane potential that activates voltage-gated proton channels, such as Hv1, to conduct an influx of protons. In addition, the ATP-driven proton pump vacuolar-type ATPase (V-ATPase) actively pumps protons in the same direction. Other cations such as K+, Na+, and Ca2+ can be transported into phagosomes via monovalent or divalent cation/H+ exchangers, or K+/Cl‒ symporter. To counterbalance the influx of the cations, Cl‒ is transported through chloride channels, including CFTR, CaCC, and voltage-gated ClCs.
Chloride is the predominant anion that regulates phagosomal oxidant production in PMN phagosomes. NADPH oxidase transfers electrons into the phagosomal lumen, thereby creating a transmembrane potential that activates voltage-gated proton channels, such as Hv1, to conduct an influx of protons. In addition, the ATP-driven proton pump vacuolar-type ATPase (V-ATPase) actively pumps protons in the same direction. Other cations such as K+, Na+, and Ca2+ can be transported into phagosomes via monovalent or divalent cation/H+ exchangers, or K+/Cl‒ symporter. To counterbalance the influx of the cations, Cl‒ is transported through chloride channels, including CFTR, CaCC, and voltage-gated ClCs.
Intraphagosomal chloride can reach as high as ∼73 mM.35 With such a phagosomal chloride level, more than 90% of the H2O2 is converted to HOCl.17,18 Therefore, the predominant form of oxidants produced by PMNs is HOCl. Highly reactive with biological targets, particularly methionine and cysteine residues in proteins, HOCl serves as a potent microbicide in PMNs.36 HOCl acts rapidly, taking only milliseconds to kill Escherichia coli,37 and very effectively, being ∼1000-fold more powerful than an equal concentration of H2O2.38 However, the activity of the MPO-H2O2-Cl system extends beyond HOCl alone and includes downstream products of reactions of HOCl with proteins to generate chloramines, aldehydes, and other agents that are themselves antimicrobial and long-lived.17,32 Furthermore, and as indicated earlier, the phagosomal environment includes other antimicrobial agents that serve as collaborators in executing microbes. Studies published long ago by Odeberg and Olsson39 catalog the multiple interactions among different granule proteins and between granule proteins and oxidants.
Chloride plays a central role in phagosomal functions
The recruitment and fusion of granules with nascent phagosomes delivers both the lumenal contents of granules and secretory vesicles but also their integral membrane proteins. Among those membrane-associated proteins are ion carriers that govern bidirectional ion flux in the organelle to support phagosomal function. Typically, whereas several different cations H+, K+, Na+, Ca2+ flux across the phagosomal membrane, the predominant anion by far is Cl‒.5,40,41
Chloride directly participates in phagosomal oxidant production
In contrast to the voltage-gated proton channel that passively conducts protons to neutralize the electrons transported by the NADPH oxidase, which is nonelectrogenic, the vacuolar-type H+-ATPase is an energy-driven proton pump that actively transports protons into the phagosomal lumen,40,42 thereby leading to a net gain of positive charges inside the lumen. Moreover, monovalent or divalent cation/H+ exchangers along with the K+/Cl‒ symporter transport K+, Na+, and Ca2+ into phagosomes and collectively augment positive charge in the lumen. These accumulated positive charges need counterions, a requirement that chloride ion fulfills. Chloride inflow occurs primarily through the CF transmembrane conductance regulator chloride channel (CFTR),5,41 calcium-activated chloride channel (CaCC),43,44 and ClC chloride channels/exchangers (ClCs)45-47 (Figure 1).
The regulation of chloride redistribution from cytoplasm into the phagosome is complex, with different signals driving each of the distinct chloride carriers and with individual chloride channels influencing the activities of each other. For example, targeted deletion of the TMEM16A gene, which encodes a calcium-activated chloride channel, from mouse intestine and airways not only eliminates Ca2+-activated Cl− currents but also abrogates CFTR-mediated Cl− secretion and completely abolishes cyclic adenosine monophosphate–activated whole cell currents, thereby suggesting that some degree of functional overlap exists between CFTR and Ca2+-dependent chloride transport.48 Based on such functional overlap, some have suggested that augmentation of the activity of members of the CaCC family represents a promising therapeutic target to compensate for CFTR defect.49
Because chloride is a prerequisite for HOCl synthesis from H2O2, its availability in phagosomes directly dictates phagosomal oxidant production, with respect to quantity and the specific product (eg, HOCl or H2O2).
Chloride directly and indirectly affects activities of proteolytic enzymes in phagosomes and oxidant microbicidal potency
Chloride is directly needed to modulate phagosomal proteolysis. Activation of dipeptidyl peptidase I or cathepsin C is chloride dependent,50 which cleaves the proenzymes of multiple serine proteases, including human neutrophil elastase, proteinase 3, cathepsin G, and neutrophil serine protease 4.51 In parallel, negatively charged electrons and chloride anion collectively limit phagosomal proton influx. The protons transported into phagosomes participate both in oxidant production (eg, H2O2, HOCl) and in pH determination. The influence of chloride on phagosomal pH is clearly demonstrated in CF PMNs, whose phagosomes are more alkaline than are those in normal PMNs.52 Phagosomal pH impacts the activity in that compartment in at least two ways.41 First, pH is a determinant for optimal activity of hydrolytic and proteolytic enzymes, thus influencing degradation of microbes within phagosomes.41,53,54 Second, phagosomal pH alters HOCl chemical equilibration in solution with hypochlorite (OCl–) and chlorine (Cl2): at pH ∼7.5, ∼50% of the compound is HOCl, and at a neutral pH, ∼80% becomes HOCl, whereas at pH 6.5, it is ∼90%.55 Because the bactericidal activity of HOCl is almost 80-fold greater than that of OCl−, chloride transport to phagosomes has a broad impact on phagosomal function. In normal PMNs, phagosomal pH is maintained at a neutral to alkalinity pH for the initial 20 to 30 minutes after phagocytosis23,33 and then slowly declines to an acidic pH, suggesting that there are two proton transport systems: one acute and one chronic. This bimodal action might serve to avoid premature inactivation of phagosomal activity in light of the strong permeability and reactivity of both HOCl and Cl2 gas.
Defects in the MPO-H2O2-Cl system compromise effective host defense and normal homeostasis
NADPH oxidase loss of function and MPO deficiency result in differential infection and inflammation diseases
Given the important contributions of the MPO-H2O2-Cl system both to host defense against infection and to resolution of the inflammatory response, one would anticipate that its dysfunction because of the absence of one or more of its constituents would undermine immune function in some clinically apparent manner. In fact, the lack of any one of the three elements of the MPO-H2O2-Cl system, namely the absence of MPO (MPO deficiency), H2O2 (chronic granulomatous disease, CGD), or Cl (CF), compromises normal host defense and resolution of inflammation, albeit not to the same extent. The clinical consequences of CGD, MPO deficiency, and CFTR differ in degree and in the specific microbial infections.
The most extreme manifestations of a defective MPO-H2O2-Cl system occur in chronic granulomatous disease
This prediction is most dramatically illustrated by chronic granulomatous disease (CGD), a genetic disorder with absent or diminished phagocyte NADPH oxidase activity. The inherited absence of any one of the five proteins known to be essential for normal oxidase activity or EROS (Essential for reactive oxygen species)56 results in PMNs unable to support oxidant-dependent microbicidal activity. As a consequence of the absence of a functional phagocyte oxidase, affected individuals have increased susceptibility to infection from some, but not all, microbes, with the most common agents being Staphylococcus aureus, Serratia marcescens, Nocardia, Aspergillus, and Burkholderia. In addition, individuals with CGD exhibit signs of exuberant and dysregulated inflammation. In fact, the initial clinical descriptions of CGD noted the predominance of granuloma and granulomatous inflammation, prompting investigators to name the disease chronic granulomatous disease. A seminal study from the Dinauer laboratory demonstrated that the exuberant inflammation in CGD was independent of infection.57 Mice that lack phagocyte oxidase activity have sustained lung inflammation compared with normal mice when challenged with sterilized hyphal cell walls from A. fumigatus.57 Numerous experimental studies and clinical observations since that publication have supported and extended the idea that resolution of the inflammatory response is defective in patients with CGD.27
The absence of MPO comprises PMN antimicrobial activity against a subset of microbes, most dramatically Candida species,12 but the defect is far less profound than that in CGD. In fact, the clinical phenotype of complete MPO deficiency is subtle and typically undetected, except in some individuals who have concomitant diabetes.58 Murine models of MPO deficiency mirror the trend seen in humans; MPO-deficient mice succumb to fungal and bacterial infections only after challenge with large inocula of organisms.4,59-61 Given the importance of synergistic interactions among antimicrobial agents in the phagosome highlighted earlier, it is not completely unexpected that CGD and MPO deficiency would have different clinical phenotypes, as the overall “microbicidal tone” in the phagosomes in the two disorders are significantly different.6,15
First, in CGD, only oxidant-independent antimicrobial activity is possible, whereas in MPO-deficient phagosomes, there is more H2O2 than in normal PMNs, because there is no MPO consumption of H2O2 to generate HOCl. Furthermore, MPO terminates the NADPH oxidase,62 and NADPH oxidase activity in stimulated MPO-deficient human PMNs is sustained,63,64 resulting in more H2O2 production. Taken together, decreased consumption and increased production result in a net increase in H2O2. Second, in the absence of HOCl production, the bioactivity of some granule proteins is increased because they are not inactivated by HOCl or its downstream products. For example, more active human neutrophil elastase is secreted from MPO-deficient PMNs than from normal PMNs, a reflection of the HOCl-mediated oxidation and inactivation of human neutrophil elastase. Last, Klebanoff65 speculated that PMNs genetically deficient in MPO may have adaptations that increase the activity of peroxidase-independent systems.
In brief, the differences between PMNs with or without oxidants are far more consequential than those between PMNs with or without MPO. The MPO-H2O2-Cl system represents the most efficient oxidant-dependent microbicidal system in PMNs but not the only system.3,36,59 To what extent, then, does the absence of sustained chloride delivery into the phagosome impair PMN host defense, both with respect to eradication of microbes and to resolution of inflammation?
CFTR loss of function leads to immunodeficiency
CF neutrophil defect leads to CF host defense failure
The availability of chloride significantly affects the bacterial killing capacity of normal PMNs. When bathed in a chloride-rich buffer, normal PMNs kill Pseudomonas aeruginosa twofold faster than they do in a chloride-free buffer (Figure 2). These data suggest that chloride availability is critical to optimal neutrophil host defense function.
![Killing of PsA by normal PMNs is chloride dependent. Normal human peripheral blood PMNs were allowed to ingest serum-opsonized PsA (20:1 PsA:PMN) in sodium gluconate chloride-free Ringer’s buffer containing 10% dialyzed human AB serum for 20 minutes at 37°C. The uningested PsAs were removed by low-speed centrifugation, and the cell pellets containing PMNs harboring PsA resuspended in Ringer’s buffer containing varied concentrations of chloride (0-127 mM) and 10% dialyzed serum. After the zero time point was sampled to determine the initial number of viable PsAs, PsA-laden PMNs were incubated with shaking at 37°C, and aliquots were taken at the indicated time points (10, 20, 30, and 40 minutes). The fraction of viable PsAs relative to those at zero time point was determined. The data are plotted on a semi-log scale relative to the viable bacteria present at zero time. [Cl], chloride concentration; K, killing rate constant.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/17/10.1182_blood.2021014699/4/m_bloodbld2021014699f2.png?Expires=1765891658&Signature=d56VbgHZWQto~IEjK7zqkjw9Gdi5b18GUIoCrPtiX-lEk64ToyFbNYYsO6y2gUQZDaDNrQDqiTrZ~DNv~DB3KUBXvgRBDnaJh48Wg8zeyeVXazYjs-P8T3mRv8O9R3mCzi1B9dNpK-41TuAdvqdSGRoU6K4ByIvBgjLfX4m6jTPZBMO8aCXdsifzFkxD5h1SqnLUtsC3a4jXF1ucVTokxwxAUh-LgK7tI0PHEUFjOE8UtWJ2fQfT~QvbBjRDNbBtC63WBGVeRYq8QM33hGdCxF8zutg-l1djk2rP0n~fZpTZsgE7hvlyrf5~Dz2XwER6fOWJBq4fs2N0-G23YLSOBA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Killing of PsA by normal PMNs is chloride dependent. Normal human peripheral blood PMNs were allowed to ingest serum-opsonized PsA (20:1 PsA:PMN) in sodium gluconate chloride-free Ringer’s buffer containing 10% dialyzed human AB serum for 20 minutes at 37°C. The uningested PsAs were removed by low-speed centrifugation, and the cell pellets containing PMNs harboring PsA resuspended in Ringer’s buffer containing varied concentrations of chloride (0-127 mM) and 10% dialyzed serum. After the zero time point was sampled to determine the initial number of viable PsAs, PsA-laden PMNs were incubated with shaking at 37°C, and aliquots were taken at the indicated time points (10, 20, 30, and 40 minutes). The fraction of viable PsAs relative to those at zero time point was determined. The data are plotted on a semi-log scale relative to the viable bacteria present at zero time. [Cl], chloride concentration; K, killing rate constant.
Killing of PsA by normal PMNs is chloride dependent. Normal human peripheral blood PMNs were allowed to ingest serum-opsonized PsA (20:1 PsA:PMN) in sodium gluconate chloride-free Ringer’s buffer containing 10% dialyzed human AB serum for 20 minutes at 37°C. The uningested PsAs were removed by low-speed centrifugation, and the cell pellets containing PMNs harboring PsA resuspended in Ringer’s buffer containing varied concentrations of chloride (0-127 mM) and 10% dialyzed serum. After the zero time point was sampled to determine the initial number of viable PsAs, PsA-laden PMNs were incubated with shaking at 37°C, and aliquots were taken at the indicated time points (10, 20, 30, and 40 minutes). The fraction of viable PsAs relative to those at zero time point was determined. The data are plotted on a semi-log scale relative to the viable bacteria present at zero time. [Cl], chloride concentration; K, killing rate constant.
The CF transmembrane conductance regulator gene (cftr) encodes CFTR, a cyclic adenosine monophosphate–activated chloride channel,66,67 whose dysfunction affects multiple organ systems, including the lung, sinuses, intestines, liver, pancreas, and vas deferens. However, chronic respiratory infection by a narrow set of bacterial pathogens, including S. aureus, Haemophilus influenzae, and P. aeruginosa,68-70 is the leading cause of morbidity and mortality in CF. Although these microbes do not typically cause pulmonary disease in the general population, more than 90% of patients with CF die from these opportunistic pathogens. CF airways are colonized by S. aureus and H. influenzae early in life. Then, P. aeruginosa progressively prevails through later childhood and adolescence. With broad-spectrum antibiotic suppression, the frequency of the respiratory tract infections caused by Burkholderia cepacia complex,71 yeast, and filamentous fungi, such as Aspergillus species, is increasing.
Locally residing cells, such as pulmonary epithelial cells and tissue macrophages, and circulating cells that migrate to lung, such as PMNs, monocytes, and eosinophils, collaborate to maintain respiratory health under normal conditions. Pulmonary epithelial cells use junctional complexes to form a tight sheet that maintains a structural barrier72 and secrete soluble immune mediators, including antimicrobial peptides such as lysozyme, defensins, and the short palate, lung, nasal epithelial clone 1, to serve as a chemical barrier to microbial invasion.73-76 The local tissue macrophages that patrol the airway and alveoli can combat and neutralize threats imposed by small numbers of microbes that may be encountered.77-79 However, when the microbial burden exceeds the capacity of local cells to respond, sentinel cells recruit circulating or marrow-reserved PMN to relocate to the site of infection, thereby amplifying the local inflammatory response.80-83 When infection is contained, macrophages efferocytose and clear the spent or apoptotic phagocytes, promoting restoration of homeostasis. Conversely, unresolved infection sustains PMN recruitment and inflammation. When PMNs are defective, as is the case with CF PMNs (see below), chronic infection and persistent neutrophilic inflammation occur.
CFTR is expressed in PMNs and preferentially stored in the secretory vesicles. Fusion of secretory vesicles with nascent phagosomes during phagocytosis integrates CFTR into phagosomal membranes, thereby providing phagosomes with a mechanism to import chloride from the cytoplasm.84,85 Using specific probes to measure phagosomal chloride and HOCl levels, we demonstrated that CFTR is a major chloride channel involved in transporting chloride into the phagosomes of normal PMNs.86-88 In the absence of CFTR, the phagosomes of CF PMNs are impermeable to chloride and do not respond to fluxes in extracellular chloride86 (Figure 3), thereby impairing phagosomal acquisition of chloride. CF human PMNs are ∼7-fold slower in transporting chloride to their phagosomes than normal PMNs, and the steady-state chloride level in phagosomes of CF PMNs is ∼2.7-fold lower than that in normal PMNs.86 Such a chloride transport defect within CF PMNs results in an incompetent chlorination and killing of phagocytosed P. aeruginosa (Figure 4),84,88,89 a microbe requiring relatively high levels of HOCl to eradicate.90 Furthermore, when P. aeruginosa becomes mucoid and forms biofilms, both PMNs and most microbicidal molecules are unable to penetrate and exert their antimicrobial action. Although oxidants such as HOCl are small enough to penetrate biofilms, their source (PMNs) has limited access to microbes in the presence of biofilms, especially in the presence of those produced by P. aeruginosa,91 and the bulk is consumed by chemical reactions with elements of the biofilms. The inability of CF PMNs to produce normal amounts of HOCl further undermines local host defense and might selectively provide a survival advantage to pathogens whose killing is predominantly HOCl dependent, including those that plague patients with CF.
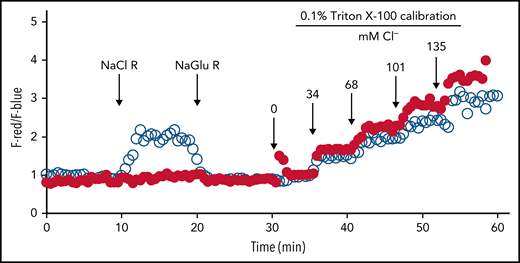
Lack of chloride influx in phagosomes of CF PMNs. Serum-opsonized zymosan particles covalently conjugated with a chloride probe (MQHA, 6-methoxyquinoline-N-6-hexanoic acid) and reference probe (TMR, tetramethylrhodamine) were ingested by normal or CF PMNs. Intraphagosomal chloride levels were continuously measured by quantitative fluorescence microscopy. The probe-laden cells were bathed in different chloride buffers in the following order: (1) sodium gluconate chloride-free Ringer’s buffer (NaGlu R), (2) sodium chloride (135 mM) Ringer’s buffer (NaCl R), and (3) chloride-free Ringer’s buffer (NaGlu R). The phagosomal chloride concentration of normal PMNs (○) rapidly respond to the change in extracellular chloride change, whereas that of CF PMN (●) had no response. At 30 minutes, a series of isoosmotic 0.1% Triton X-100 solutions containing varied chloride concentrations (0-135 mM) was introduced sequentially to calibrate the system at 6-minute intervals. Reprinted from reference 86.
Lack of chloride influx in phagosomes of CF PMNs. Serum-opsonized zymosan particles covalently conjugated with a chloride probe (MQHA, 6-methoxyquinoline-N-6-hexanoic acid) and reference probe (TMR, tetramethylrhodamine) were ingested by normal or CF PMNs. Intraphagosomal chloride levels were continuously measured by quantitative fluorescence microscopy. The probe-laden cells were bathed in different chloride buffers in the following order: (1) sodium gluconate chloride-free Ringer’s buffer (NaGlu R), (2) sodium chloride (135 mM) Ringer’s buffer (NaCl R), and (3) chloride-free Ringer’s buffer (NaGlu R). The phagosomal chloride concentration of normal PMNs (○) rapidly respond to the change in extracellular chloride change, whereas that of CF PMN (●) had no response. At 30 minutes, a series of isoosmotic 0.1% Triton X-100 solutions containing varied chloride concentrations (0-135 mM) was introduced sequentially to calibrate the system at 6-minute intervals. Reprinted from reference 86.
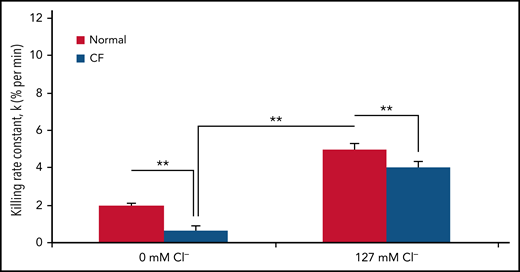
CF neutrophil killing of P. aeruginosa is impaired and further compromised in a chloride-poor environment. The rate of PsA killing, indicated by the first-order rate constant (% per minute), by normal (solid bars) or CF (open bars) PMNs in chloride-poor medium (0 mM chloride) or chloride-rich medium (127 mM chloride). Error bars represent standard error of the mean (n = 5 donors each). **P ≤ .05, for comparisons of the indicated means by Student t test. Reprinted from reference 89.
CF neutrophil killing of P. aeruginosa is impaired and further compromised in a chloride-poor environment. The rate of PsA killing, indicated by the first-order rate constant (% per minute), by normal (solid bars) or CF (open bars) PMNs in chloride-poor medium (0 mM chloride) or chloride-rich medium (127 mM chloride). Error bars represent standard error of the mean (n = 5 donors each). **P ≤ .05, for comparisons of the indicated means by Student t test. Reprinted from reference 89.
The prevailing theory of CF pathogenesis asserts that the CF airway epithelial cell defect impairs chloride secretion to the airway lumen, reduces water retention in the surface liquid, and compromises the mucociliary clearance capacity, hence creating a static milieu that invites microbial colonization.92 Whereas this theory explains well the early stages of CF whereby deposited microbes colonize the CF lung, it does not explain the selection of microbes that predominate infections seen in CF; the failure of mucociliary transport to clear entrapped bacteria should do so indiscriminately without regard to the particular bacterial species. In contrast to other airway obstructive diseases, such as chronic obstructive pulmonary disease, primary ciliary dyskinesia, and non-CF bronchiectasis, CF lung disease exhibits a characteristic infection profile, nature of inflammation, and pattern of obstruction. Specifically, infections with S. aureus and Pseudomonas species predominate, altogether occupying ∼70% to 90% of all CF cases.93 CF lung inflammation is predominantly neutrophilic and obstinately persistent, and CF lung obstruction is very purulent and occurs primarily in the small airways. Collectively, these pathologic presentations clearly implicate PMNs as major effectors of CF pathology, as defective PMN support uncontrolled infection and inflammation. Furthermore, the formation of neutrophil extracellular traps and necrosis release large amounts of PMN DNA and debris, inevitably forming pus that plugs airways.94
If the CF airway epithelial cell defect alone drove CF pulmonary disease, replacement of the defective airway epithelial cells would completely eradicate pulmonary disease in CF. Lung transplantation is an established procedure to treat patients with a variety of end-stage lung diseases, including CF, and improves many aspects of pulmonary function. However, despite aggressive measures to eradicate preoperative sinus infection, Pseudomonas remains the most common cause of bronchopulmonary infections in patients with CF after lung transplantation.95 Data from one transplantation center indicate that CF lung transplants are more susceptible to Pseudomonas infection than are individuals with non-CF lung transplants.96 Furthermore, examinations of the pathology in failed lung transplants from patients with CF undergoing a second transplantation reveal that the healthy lungs previously implanted in patients with CF resembled CF-like lungs, displaying the infection, inflammation, and small airway obstruction typical of CF pathology of (G. Wang and V.G. Valentine, personal observations, 2006). Hence, providing patients with CF with healthy lungs clearly improves many but not all features of their pulmonary disability, suggesting that the inherently defective CF immune system contributes to the onset, development, and progression of CF lung disease.
The beneficial responses to some clinical interventions that target PMN-dependent events support the concept that PMN primarily promote CF disease and are not simply inadvertent contributors that are secondary to infection. Some of the beneficial effects of DNase treatment have been attributed to disrupting neutrophil extracellular trap formation,94,97 and CFTR modulators such as ivacaftor have been linked to decreasing interleukin-8–mediated recruitment of PMNs into lung,98 correcting intrinsic defects in PMNs,99 and improving Cl efflux in monocytes.100
Consistent with this perspective is the observation that CFTR modulation therapy alleviates CF lung disease but falls short in overcoming CF lung infection and inflammation,101 because the drugs are not designed to target PMNs and macrophages that were recruited to the lung. It is conceivable that after their migration to the airway lumen, marrow-derived immune cells may be less responsive to CFTR modulation because of suboptimal drug levels, ineffective drug dynamics in the airways, or both. In support of an important role for myeloid cells in CF, a monocyte-specific knockout of CFTR primes mice for more severe experimental colitis, whereas bone marrow transplantation reverses the monocyte defect and improves CF in a murine model.102 Furthermore, studies that use reciprocal bone marrow transplantation between wild-type and pan-CF mice demonstrate that the genotype of donors determines the phenotype of lung response to infection.103,104 Specifically, immune reconstitution of CF mice by wild-type marrow cells rescues the mice from hyperinfection and hyperinflammation after bacterial challenge. Of note, bone marrow transplantation reconstitutes the entire immune system, which includes lung-recruited PMNs and monocytes/macrophages.
CF neutrophil defect causes neutrophilic inflammation
The elimination of invading pathogens by PMNs is the most effective way to terminate inflammation,105 and the unresolved infection and resulting stimulation in CF fuels sustained inflammation and secondary tissue damage. Experimental models of infection demonstrate that myeloid CFTR-knockout mice exhibit defective clearance of bacteria and resolution of inflammation.88,106,107 Persistent neutrophilic inflammation characterizes CF inflammatory disease, which starts at an early age in the lungs of patients with CF.108 Unlike CGD, patients with CF rarely develop granulomas but instead arrest the inflammatory response at the acute neutrophilic phase. Mice with CFTR loss-of-function in myeloid innate immune cells exhibit prolonged neutrophilic inflammation on zymosan challenge.107 Furthermore, data demonstrate that the defective neutrophils per se are a key factor leading to the neutrophilic inflammation.
In addition to the neutrophilic inflammation in the lung, neutrophilic inflammation is also a hallmark of CF intestinal disease.109-111 Clinically, endoscopic lavage of CF small intestines showed increased levels of inflammatory markers in the lumen.112 Video-equipped capsule endoscopy reveals obvious intestinal morphologic abnormalities including edema, erythema, mucosal breaks, and ulcerations in the jejunum and ileum in ∼60% of patients with CF. Fecal neutrophil marker proteins are significantly elevated.113 Strikingly, there is an increased prevalence of Crohn’s disease in patients with CF, up to 12.5-fold greater than in the general population.114 Celiac disease is a destructive autoimmune condition of the small intestinal mucosa that is also more prevalent in CF.115,116 Histopathologic examination of CF intestines reveals neutrophilic inflammation in the submucosal region. These clinical and experimental observations suggest that CF neutrophils are actively involved in CF pathogenesis.
Neutrophilic inflammation leads to organ damage and failure
Neutrophilic inflammation occurs in all epithelial-lined CF-targeted organs. The excessive numbers of recruited PMNs suffer a necrotic death, characterized morphologically by nuclear and organellar swelling (oncosis) and plasma membrane rupture.117 The decomposing PMNs release their intracellular contents, including the granule stores of proteases, oxidases, lipases, and other toxic molecules, which in turn damage the local tissues. Of all active agents released from dying PMNs, the serine protease neutrophil elastase exerts the most damage. It hydrolyzes many proteins in addition to elastin, induces increased mucin expression and secretion, impairs ciliary motility, and inhibits several innate immune functions by digesting opsonins and opsonin receptors, degrading innate immune proteins such as lactoferrin, and inhibiting macrophage phagocytosis.118 In CF lungs, the amount of free neutrophil elastase in the airway highly correlates with lung dysfunction (FEV1).119 As in CF lung disease, neutrophilic inflammation is a prominent feature of CF liver disease, which occurs in∼40% in patients with CF and accounts for 2% to 5% of overall CF mortality.120 CFTR is expressed on the apical surface of cholangiocytes and gallbladder epithelia.121 Defective CFTR impairs secretion of Cl− and leads to viscous bile with reduced flow and alkalinity. Stagnation of the abnormal bile prompts hepatic neutrophilic inflammation, which causes bile duct plugging, hepatic injury, and fibrosis.122 CF also targets the pancreas and does so early in the disease, which prompted initial observers to call CF the “cystic fibrosis of the pancreas.” Pancreatic damage results from severe inflammation and secretion of viscous, protein rich fluid, leading to obstruction of ducts, calcification, destruction of acini, pancreatic cyst formation, and eventually fibrosis.123 Therefore, excessive neutrophilic inflammation constitutes a key factor in CF disease development and progression that culminates in organ failure.
Concluding remarks
Oxidant production and the MPO-H2O2-Cl system are critical contributors to two physiologically important PMN activities, namely antimicrobial host defense and resolution of the inflammatory response. Defective operation of the MPO-H2O2-Cl system because of a deficiency in any component has significant consequences and a range of clinical manifestations that compromise the health of affected individuals. Mutations in CFTR compromise normal chloride influx into the phagosomes of CF PMNs, undermine the activity of the MPO-H2O2-Cl system, and culminate in defective PMN function, with respect both to host defense and resolution of the inflammatory response. An understanding of this pathophysiology and its contribution to the dysfunction of multiple organ system in CF provides a foundation for novel therapies to target defective CFTR in its many clinical manifestations.
Acknowledgments
The authors apologize to the authors of many outstanding publications that had to be omitted because of the limitation in references. The authors thank their former and current team members who contributed to the CF neutrophil research partially presented in this article.
This work was supported by grants from the National Heart, Lung and Blood Institute (HL150370 to G.W.), the National Institute of Allergy and Infectious Diseases (AI140088 to G.W. and AI132335 and AI116546 to W.M.N.), and the Cystic Fibrosis Foundation (WANG18I0 to G.W.).
Authorship
Contribution: G.W. and W.M.N. planned, wrote, and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Guoshun Wang, Department of Microbiology, Immunology, and Parasitology, Louisiana State University Health Sciences Center, New Orleans, LA 70112; e-mail: gwang@lsuhsc.edu; and William M. Nauseef, Inflammation Program, Department of Medicine, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, 501 EMRB, Iowa City, IA 52240; e-mail: william-nauseef@uiowa.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal