

Key Points
Platelet-derived CXCL12 activates platelets through Btk contributing to collagen-dependent arterial thrombosis.
The CCL5-derived peptide i[VREY]4 inhibits CXCL12 engaging CXCR4 on activated platelets and curbs thrombosis without causing leukocytosis.

Abstract
The prevention and treatment of arterial thrombosis continue to be clinically challenging, and understanding the relevant molecular mechanisms in detail may facilitate the quest to identify novel targets and therapeutic approaches that improve protection from ischemic and bleeding events. The chemokine CXCL12 augments collagen-induced platelet aggregation by activating its receptor CXCR4. Here we show that inhibition of CXCR4 attenuates platelet aggregation induced by collagen or human plaque homogenate under static and arterial flow conditions by antagonizing the action of platelet-secreted CXCL12. We further show that platelet-specific CXCL12 deficiency in mice limits arterial thrombosis by affecting thrombus growth and stability without increasing tail bleeding time. Accordingly, neointimal lesion formation after carotid artery injury was attenuated in these mice. Mechanistically, CXCL12 activated via CXCR4 a signaling cascade involving Bruton’s tyrosine kinase (Btk) that led to integrin αIIbβ3 activation, platelet aggregation, and granule release. The heterodimeric interaction between CXCL12 and CCL5 can inhibit CXCL12-mediated effects as mimicked by CCL5-derived peptides such as [VREY]4. An improved variant of this peptide, i[VREY]4, binds to CXCL12 in a complex with CXCR4 on the surface of activated platelets, thereby inhibiting Btk activation and preventing platelet CXCL12-dependent arterial thrombosis. In contrast to standard antiplatelet therapies such as aspirin or P2Y12 inhibition, i[VREY]4 reduced CXCL12-induced platelet aggregation and yet did not prolong in vitro bleeding time. We provide evidence that platelet-derived CXCL12 is involved in arterial thrombosis and can be specifically targeted by peptides that harbor potential therapeutic value against atherothrombosis.
Introduction
Arterial thrombosis is a major health care challenge giving rise to myocardial infarction and stroke as leading causes of cardiovascular mortality. As the underlying pathology, atherosclerotic plaques can rupture, exposing collagens, activating platelets, and triggering the coagulation cascade to form a clot and block arterial blood flow.1 Therefore, heparin and platelet inhibitors have become standard as first-line treatment during acute events, followed by dual antiplatelet therapy. However, our understanding of the platelet machinery that mediates this pathology is incomplete, and bleeding complications encountered with current therapies prompt an unmet clinical need to extend therapeutic options.
Platelets play a central role in arterial thrombosis and express chemokine receptors, namely CCR4 interacting with CCL17 or CCL22 and CXCR4 interacting with CXCL12, which can mediate platelet activation.2-5 The effect of the CXCL12–CXCR4 axis on platelet activation has been studied in the greatest detail. Cooperative effects on platelet aggregation induced by the CXCL12–CXCR4 axis have been observed when platelets are costimulated with different agonists such as adenosine 5′-diphosphate (ADP), thrombin, or collagen at low doses.3-9
The details of CXCL12/CXCR4–dependent platelet activation are less well understood than glycoprotein VI (GPVI)-dependent signaling pathways. Phosphatidylinositol 3-kinase (PI3K), an as yet unspecified tyrosine kinase, Akt, and MAPK are known to be involved.3,8,9 Collagen/GPVI signaling involves a Syk-dependent signaling cascade in which a LAT signalosome consisting of adaptor, effector, and kinase proteins, including PI3K and Btk, lead to PLCγ2 activation, Ca2+ release, and integrin activation. On the other side, Additionally, PI3K additionally activates Akt via p38 MAPK.10
Most CXCL12 in plasma is not derived from hematopoietic cells, including platelets, but rather from tissue-derived cells.11 However, platelets can store CXCL12, which is released upon activation and may thus play a primarily localized role when deposited on neighboring cells such as other platelets, endothelial cells, or matrix surfaces exposed upon vascular injury.12-16 Numerous stimuli, namely GPVI agonists such as collagen, which become exposed by endothelial denudation and are prothrombotic components of atherosclerotic plaques, can activate platelets to trigger chemokine release.17-19 CXCL12 released by activated platelets feeds into an autocrine forward loop by activating platelets via CXCR4.6 However, whether this mechanism is relevant to arterial thrombosis has not been studied or therapeutically evaluated. CXCL12 can form heterodimers with other inflammatory mediators (eg, CCL5, galectin-3) that functionally inhibit CXCL12.20,21 Targeting CXCL12 in platelet activation through this concept may represent a promising new therapeutic modality.
Methods
Additional details on Methods are presented in the supplemental Materials and Methods (available on the Blood Web site). Informed consent was obtained, as per the Declaration of Helsinki.
Mice
All experimental procedures were performed in agreement with the German Animal Welfare Legislation, reviewed and approved by the local authorities (Regierung von Oberbayern). C57BL/6J mice were from Janvier, B6.129P2-Apoetm1Unc/J mice were from Charles River Laboratories, and Pf4-Cre mice were from The Jackson Laboratory. Cxcl12flox/flox mice were generated in-house.20CreErtwt/wt Cxcr4flox/flox and CreErttg/wt Cxcr4flox/flox mice were generated in-house as previously described.22
Ferric chloride–induced arterial thrombosis
Mice were given intraperitoneal anesthesia (ie, medetomidine 0.5 mg/kg, midazolam 5 mg/kg, fentanyl 0.05 mg/kg), and injected with 100 µg i[VREY]4 or an equimolar amount of VREY control (20 µg) in phosphate-buffered saline (PBS) or PBS alone (200 µL each) 1 hour before the procedure. Carotid artery thrombosis was induced by 10% ferric chloride (FeCl3) and blood flow monitored by Doppler sonography, as detailed in the supplement.
Flow cytometry analysis
Mouse platelets were gated by CD41 (MWReg30; Novus Biologicals), and activation by collagen was analyzed by detecting P-selectin (Wug.E9-FITC mAb, D200; Emfret Analytics) and αIIbβ3 (JON/A-PE mAb, D200; Emfret Analytics). Permeabilized platelets were reacted with a phycoerythrin-labeled anti-Btk Phospho (Tyr223) antibody (clone A16128B; BioLegend). For human platelets, whole blood was diluted 1:1 with saline and activated as detailed in the legend for Figure 6C-F and supplement (see “Flow cytometric analysis of CXCR4 internalization, CXCL12 binding/expression and i[VREY]4 binding”).
CXCR4 and CXCL12 on the surface of human platelets were analyzed by using anti-CXCR4 (12G5; R&D Systems) or anti-CXCL12 (K15C-Star Red, Merck; clone 79018-FITC, R&D Systems) in human blood diluted 1:1 with PBS, as detailed in the supplement. Platelet activation of human platelets was assessed by PAC-1 (activated αIIbβ3; BD Biosciences) and P-selectin antibody (AK-4; BD Biosciences) staining with and without Btk inhibition (0.1 µM remibrutinib, 30 minutes at 37°C) before stimulation with combinations of recombinant CXCL12 and CRP-XL.
Binding of a biotinylated form of i[VREY]4 (i[VREY]4-biot) to human or mouse platelets was detected with streptavidin–fluorescein isothiocyanate and analyzed by using flow cytometry (Vector Laboratories). Blood from CreErtwt/wtCxcr4flox/flox (wild type) and CreErttg/wtCxcr4flox/flox (CXCR4 knockout) mice was used 4 weeks after tamoxifen injection. After red blood cell lysis, blood was stained with anti-CD41 (MWReg30; ExBio), anti-CD45 (30F11; Invitrogen), and anti-Ly6G (1A8; BioLegend) antibodies, and platelet–neutrophil complexes were defined as CD45+Ly6G+CD41+ cells.
Ex vivo thrombus formation of mouse blood
Multiparameter assessment of murine blood was performed as described.23 Details are provided in the supplemental Materials and Methods.
Collection and processing of human atherosclerotic plaques
Atherosclerotic plaques were collected from carotid endarterectomies and processed to a homogenate, as previously described.24
Multiple electrode aggregometry
Human platelet aggregation in blood anticoagulated with hirudin was determined by multiple electrode aggregometry (MEA) using the Multiplate device, as reported previously,25,26 for 15 minutes. Blood was treated with Horm collagen (from equine tendon; Takeda), human plaque homogenate, recombinant CXCL12, CCL5 or CCL1, and pretreated with inhibitors as detailed in the respective figure legends.
Statistical analysis
Data are expressed as mean ± standard deviation and analyzed by using GraphPad Prism version 8 (GraphPad Software). Inhibitor and concurrent controls from the same donor were compared by using the paired t test. For unpaired data, when D’Agostino-Pearson omnibus normality test indicated a Gaussian distribution, a t test for side-by-side comparisons or analysis of variance with posttests was used, as indicated. Otherwise, Mann-Whitney U tests were used.
Results
Platelet-derived CXCL12 promotes arterial thrombosis
To evaluate the relevance of platelet-derived CXCL12 in vivo, we generated mice with a specific deletion of Cxcl12 in the megakaryocyte lineage (Cxcl12Δplt/Δplt) by crossing Pf4-Cre+ and Cxcl12fl/fl mice20 in an Apoe−/− background. CXCL12 plasma levels did not differ between Pf4-Cre+Cxcl12Δplt/Δplt mice and Cxcl12wtlwt littermates (supplemental Table 1), confirming that under physiological, steady-state conditions, neither platelets nor other hematopoietic cells appreciably contribute to circulating CXCL12 levels.11 Body weight and blood cell counts did not differ. In a model of FeCl3-induced arterial thrombosis,27 occlusion occurred significantly later in Cxcl12Δplt/Δplt mice than in Cxcl12wtlwt mice (Figure 1A). Likewise, thrombus growth and stability were impeded in Cxcl12Δplt/Δplt mice (Figure 1B).
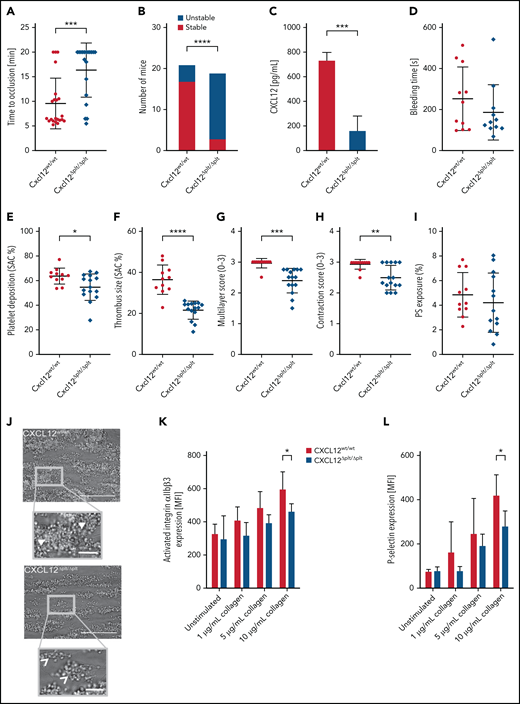
Platelet-derived CXCL12 promotes arterial thrombosis. (A-B) Thrombus formation was induced by FeCl3 in the carotid artery of ApoE−/− mice (n = 22). The time to occlusion (A) was measured by using Doppler sonography, and thrombi were classified into “stable” and “unstable” (B) as specified in the Methods. (C) Isolated mouse blood was activated with collagen (10 µg/mL), and the concentration of CXCL12 from the releasate was determined by enzyme-linked immunosorbent assay (n = 3). (D) Tail bleeding time was assessed (n = 11). (E-I) Multiparameter analysis of thrombus formation in a collagen-coated flow chamber perfused with murine whole blood (1000 s−1): platelet deposition (E), thrombus size (F), thrombus multilayer score (G), thrombus contraction score (H), and phosphatidylserine (PS) exposure (I) were assessed by Annexin V staining. (J) Representative micrographs (n = 11-15); note that Cxcl12wtlwt mice form large and contracted thrombi, in which individual platelets are barely recognizable (closed arrow heads), whereas Cxcl12Δplt/Δplt mice tend to generate smaller less contracted thrombi featuring clearly distinguishable individual platelets (open arrow head); scale bar overview, 50 µm; scale bar inlet, 10 µm. (K-L) Platelet activation by collagen (1, 5, and 10 µg/mL) was analyzed by upregulation of activated αIIbβ3 (K) and P-selectin (L) by flow cytometry (n = 6). Data represent mean ± standard deviation from the indicated numbers of independent experiments or mice. *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001, as analyzed by using the Mann-Whitney U test (panels A and D), Fischer’s exact test (panel B), and unpaired t tests (panels C, E-I, K, and L). MFI, mean fluorescence intensity; SAC, surface area coverage.
Platelet-derived CXCL12 promotes arterial thrombosis. (A-B) Thrombus formation was induced by FeCl3 in the carotid artery of ApoE−/− mice (n = 22). The time to occlusion (A) was measured by using Doppler sonography, and thrombi were classified into “stable” and “unstable” (B) as specified in the Methods. (C) Isolated mouse blood was activated with collagen (10 µg/mL), and the concentration of CXCL12 from the releasate was determined by enzyme-linked immunosorbent assay (n = 3). (D) Tail bleeding time was assessed (n = 11). (E-I) Multiparameter analysis of thrombus formation in a collagen-coated flow chamber perfused with murine whole blood (1000 s−1): platelet deposition (E), thrombus size (F), thrombus multilayer score (G), thrombus contraction score (H), and phosphatidylserine (PS) exposure (I) were assessed by Annexin V staining. (J) Representative micrographs (n = 11-15); note that Cxcl12wtlwt mice form large and contracted thrombi, in which individual platelets are barely recognizable (closed arrow heads), whereas Cxcl12Δplt/Δplt mice tend to generate smaller less contracted thrombi featuring clearly distinguishable individual platelets (open arrow head); scale bar overview, 50 µm; scale bar inlet, 10 µm. (K-L) Platelet activation by collagen (1, 5, and 10 µg/mL) was analyzed by upregulation of activated αIIbβ3 (K) and P-selectin (L) by flow cytometry (n = 6). Data represent mean ± standard deviation from the indicated numbers of independent experiments or mice. *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001, as analyzed by using the Mann-Whitney U test (panels A and D), Fischer’s exact test (panel B), and unpaired t tests (panels C, E-I, K, and L). MFI, mean fluorescence intensity; SAC, surface area coverage.
When blood was activated with collagen, a substantial release of CXCL12 was observed in Cxcl12wtlwt mice but not in Cxcl12Δplt/Δplt mice, validating our model (Figure 1C).28 Tail bleeding times in Cxcl12Δplt/Δplt and Cxcl12wtlwt mice were comparable. Therefore, we could exclude a critical role of CXCL12 in primary hemostasis (Figure 1D).
To substantiate our findings ex vivo, we perfused whole blood from Cxcl12Δplt/Δplt mice and Cxcl12wtlw mice through collagen-coated microfluidics chambers.29 A multiparameter assessment revealed the presence of smaller thrombi, as evident by a decrease in platelet deposition, thrombus size, multilayer score, and thrombus contraction score (Figure 1E-H), the latter indicating decreased stability of thrombi from Cxcl12Δplt/Δplt blood (Figure 1J). In line with reduced stability, the more pronounced reduction of thrombus size than of platelet deposition suggests an αIIbβ3-integrin–dependent process of platelet activation by CXCL12.29 The proportion of procoagulant, phosphatidylserine-exposing platelets did not differ between genotypes (Figure 1I) or by fluorescence-activated cell sorting analysis (data not shown).
After FeCl3-induced injury of the left carotid artery, mice were put on a Western diet (WD) for 4 weeks, leading to the formation of neointimal lesions, which appeared to be reduced in size, albeit not significantly, in Cxcl12Δplt/Δplt mice vs Cxcl12wtlwt mice (supplemental Figure 1A). Platelet–neutrophil complexes did not differ between genotypes in mice fed a chow diet (supplemental Figure 1B). In line with previous findings,30 however, WD for 4 weeks increased circulating platelet–neutrophil complexes in Cxcl12wtlwt mice compared with Cxcl12Δplt/Δplt mice. Because the size of atherosclerotic plaques in the aortic root was unaltered (supplemental Figure 1C), we surmised that platelet-derived CXCL12 does not play a crucial role in early-stage atherosclerosis. CXCL12 plasma concentrations in mice on chow or WD were comparable to those from previous studies and did not differ between genotypes (supplemental Tables 1 and 2), confirming the minor contribution of platelet CXCL12 to systemic levels.11
Upon collagen stimulation of blood, activation of integrin αIIbβ3 (Figure 1K) and P-selectin expression (Figure 1L) were attenuated in Cxcl12Δplt/Δplt mice vs that in Cxcl12wtlwt mice. Both receptors contribute to the formation of platelet–neutrophil complexes. Therefore, the lower abundance of platelet–neutrophil complexes in Cxcl12Δplt/Δplt mice detectable under WD but not under a chow diet (supplemental Figure 1B) likely reflects a reduction in platelet activation or local CXCL12 availability in the context of hypercholesterolemia.31 Our in vivo findings indicate that platelet-derived CXCL12 plays an important role in atherothrombosis without affecting hemostasis or early atherogenesis.
Human platelet aggregation and thrombus formation stimulated by collagen and plaque involves a CXCL12–CXCR4 feedback loop
After plaque rupture, fibrillar collagen is crucial for platelet activation and arterial thrombosis, prompting antagonists of its receptor GPVI as a therapeutic option.32,33 Platelet aggregation induced by collagen or human plaque under static conditions in MEA was reduced by the CXCR4 antagonist AMD3465 (Figure 2A-B). Similarly, the volume of nascent thrombi that form when a plaque-coated flow chamber was perfused with human blood was diminished by AMD3465 (Figure 2C; supplemental Videos 1-3). This is consistent with a positive feedback-loop via the CXCR4–CXCL12 axis.7 The CXCL12 concentration in our plaque homogenate was 21 ± 10 ng/mL, which, when diluted to 83 pg/mL for MEA, would be too low to trigger platelet aggregation. CXCL12 was present in platelets and released by collagen (Figure 2D-E). At ≥100 ng/mL concentrations, CXCL12 induced platelet aggregation (Figure 2F). CXCL12 cooperated with low-dose collagen to induce platelet aggregation via CXCR4 (Figure 2G), explaining why AMD3465 inhibits the plaque-induced response.
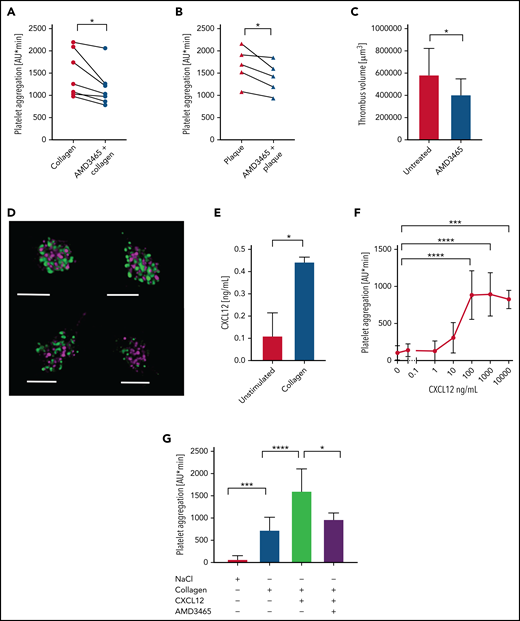
The CXCL12–CXCR4 axis functions as a positive feedback loop in human platelet activation. Platelet aggregation was assessed by MEA in human blood activated by collagen (0.2 μg/mL) (A) or human plaque homogenate (B). CXCR4 was inhibited by 100 nM AMD3465 (n = 5-8). (C) Thrombus formation was induced by perfusion (600 s−1) of human blood, preincubated with PBS or 1 μM AMD3465, in a plaque-coated flow chamber and thrombus volume determined by confocal microscopy (n = 7). (D) CXCL12 was visualized in resting human platelets that were permeabilized and double-stained with antibodies against CXCL12 (purple) and CXCL4 antibody (green) by stimulated emission depletion (STED) microscopy (Leica SP8; scale bar, 2 μm). (E) CXCL12 release from isolated human platelets after activation with collagen (5 μg/mL) was determined by enzyme-linked immunosorbent assay (n = 3). (F-G) Platelet aggregation was assessed by MEA of human blood incubated with different concentrations of recombinant CXCL12 (n = 5-10) (F) or combinations of collagen (0.1 μg/mL), recombinant CXCL12 (0.1 μg/mL), and AMD3465 (100 nM) as indicated (n = 6-10) (G). Data represent mean ± standard deviation from the indicated numbers of independent experiments. *P ≤ .05, ***P ≤ .001, ****P ≤ .0001, as analyzed by paired t test (panels A and B), unpaired t test (panels C and E), and one-way analysis of variance with Tukey’s multiple comparison test (panels F and G). AU, arbitrary units.
The CXCL12–CXCR4 axis functions as a positive feedback loop in human platelet activation. Platelet aggregation was assessed by MEA in human blood activated by collagen (0.2 μg/mL) (A) or human plaque homogenate (B). CXCR4 was inhibited by 100 nM AMD3465 (n = 5-8). (C) Thrombus formation was induced by perfusion (600 s−1) of human blood, preincubated with PBS or 1 μM AMD3465, in a plaque-coated flow chamber and thrombus volume determined by confocal microscopy (n = 7). (D) CXCL12 was visualized in resting human platelets that were permeabilized and double-stained with antibodies against CXCL12 (purple) and CXCL4 antibody (green) by stimulated emission depletion (STED) microscopy (Leica SP8; scale bar, 2 μm). (E) CXCL12 release from isolated human platelets after activation with collagen (5 μg/mL) was determined by enzyme-linked immunosorbent assay (n = 3). (F-G) Platelet aggregation was assessed by MEA of human blood incubated with different concentrations of recombinant CXCL12 (n = 5-10) (F) or combinations of collagen (0.1 μg/mL), recombinant CXCL12 (0.1 μg/mL), and AMD3465 (100 nM) as indicated (n = 6-10) (G). Data represent mean ± standard deviation from the indicated numbers of independent experiments. *P ≤ .05, ***P ≤ .001, ****P ≤ .0001, as analyzed by paired t test (panels A and B), unpaired t test (panels C and E), and one-way analysis of variance with Tukey’s multiple comparison test (panels F and G). AU, arbitrary units.
A peptide-based CXCL12 inhibitor prevents arterial thrombosis
Previously, we discovered and characterized chemokine–chemokine heterodimers that can enhance or inhibit chemokine function.20 Using structure-based evidence of these novel chemokine interactions, we designed peptides from the contact regions, thereby modulating chemokine activity. Here, we confirmed that CCL5 effectively inhibits CXCL12-induced platelet aggregation (supplemental Figure 2A) and that inhibitory effects of CCL5 on CXCL12 by CXC-type heterodimer formation can be mimicked by scaffolded peptides from the CCL5 C-terminal α-helix (54-68) that harbors the eponymous residues VREY (…EKKWVREYINSLEMS). We linked four VREY molecules on a scaffold to promote helix formation and termed this construct [VREY]4 (supplemental Figure 2B). To enhance helix structure and stability, we generated a new scaffold version termed i[VREY]4, a biotinylated form (i[VREY]4-biot) and a nonscaffold VREY control (supplemental Figure 2C-E).
Ligand blots qualitatively showed that i[VREY]4 and [VREY]4 but not VREY control interact with CXCL12 (supplemental Figure 3A). Surface plasmon resonance kinetics revealed that CXCL12 binds with nanomolar affinity to i[VREY]4-biot immobilized on a neutravidin-coated sensor chip (Figure 3A). Using 15N-labeled CXCL12, HSQC nuclear magnetic resonance (NMR) titrations with i[VREY]4 showed that i[VREY]4 interacts with CXCL12 with an affinity in the micromolar range (Figure 3B; supplemental Figure 3B-C). NMR titration plots could be fit with a single exponential (supplemental Figure 3D), indicating the presence of a 2-state (free and bound CXCL12) equilibrium process. These results are in agreement with a direct binding of i[VREY]4 to CXCL12 monomers.34 However, whereas some resonances follow monomer-to-dimer shift patterns, others do not.
![i[VREY]4, a CCL5-mimicking peptide, binds to CXCL12. (A) Binding kinetics of CXCL12 to i[VREY]4 by surface plasmon resonance. i[VREY]4-biot was immobilized onto a neutravidin-conjugated C1 sensor chip (914 RU), and CXCL12 was injected at 62.5 ng/mL, 125 ng/mL, 250 ng/mL, 500 ng/mL, and 1000 ng/mL. Red traces represent the single-site fit to the raw data (blue). Kinetic parameters of 3 independent experiments are indicated as mean ± standard deviation. (B) Expansions of 15N HSQC spectra are shown for 15N-labeled CXCL12 in the absence (red peaks) and presence of i[VREY]4 at concentrations of 20 µM (green peaks), 40 µM (purple peaks), and 200 µM (blue peaks). (C) The interaction of endogenous CXCL12 with i[VREY]4-biot (1 µM) in human blood was quantified on platelets by proximity ligation with DuoLink by flow cytometry (C) and visualized by confocal microscopy on platelets (scale bar, 2 µm) (n = 3) (D). Data represent mean ± standard deviation from the indicated numbers of independent experiments. **P ≤ .01, ***P ≤ .001 as analyzed by one-way analysis of variance with Dunnett’s multiple comparison test (panel C). MFI, mean fluorescence intensity.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/17/10.1182_blood.2020010140/4/m_bloodbld2020010140f3.png?Expires=1765884507&Signature=AG4E~sR5PCjmo4br-qdz7LaEop924gIa51ldqWYEH8QUBwCR95MrSokYrm4-b5SWdmEDTiPhVeSFSLdB5FZJC-a6d3Ai0cVyOZUHLOwuvE~6JU8qcjicbJ25yqD1LeSJFh8gr-c9AeAjZdEanwfvBqTvtnD28Na6i5VFskYf-E5y6hMFGh6BGhJX2ULsiFWUwg71b2MPlJUlo4u4~tE3XAhycJWSaFTA7~VMz~UxcqVQ8TOg5npi4B1HRM6pZ8RKqrHJObwlxGg-oAfvn5210ZEc0iIXX2K4clzSaYXPmnd0humO4zFl7~UXNTPl7SIMDstKxdmh5UesX~mHpQUREg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
i[VREY]4, a CCL5-mimicking peptide, binds to CXCL12. (A) Binding kinetics of CXCL12 to i[VREY]4 by surface plasmon resonance. i[VREY]4-biot was immobilized onto a neutravidin-conjugated C1 sensor chip (914 RU), and CXCL12 was injected at 62.5 ng/mL, 125 ng/mL, 250 ng/mL, 500 ng/mL, and 1000 ng/mL. Red traces represent the single-site fit to the raw data (blue). Kinetic parameters of 3 independent experiments are indicated as mean ± standard deviation. (B) Expansions of 15N HSQC spectra are shown for 15N-labeled CXCL12 in the absence (red peaks) and presence of i[VREY]4 at concentrations of 20 µM (green peaks), 40 µM (purple peaks), and 200 µM (blue peaks). (C) The interaction of endogenous CXCL12 with i[VREY]4-biot (1 µM) in human blood was quantified on platelets by proximity ligation with DuoLink by flow cytometry (C) and visualized by confocal microscopy on platelets (scale bar, 2 µm) (n = 3) (D). Data represent mean ± standard deviation from the indicated numbers of independent experiments. **P ≤ .01, ***P ≤ .001 as analyzed by one-way analysis of variance with Dunnett’s multiple comparison test (panel C). MFI, mean fluorescence intensity.
i[VREY]4, a CCL5-mimicking peptide, binds to CXCL12. (A) Binding kinetics of CXCL12 to i[VREY]4 by surface plasmon resonance. i[VREY]4-biot was immobilized onto a neutravidin-conjugated C1 sensor chip (914 RU), and CXCL12 was injected at 62.5 ng/mL, 125 ng/mL, 250 ng/mL, 500 ng/mL, and 1000 ng/mL. Red traces represent the single-site fit to the raw data (blue). Kinetic parameters of 3 independent experiments are indicated as mean ± standard deviation. (B) Expansions of 15N HSQC spectra are shown for 15N-labeled CXCL12 in the absence (red peaks) and presence of i[VREY]4 at concentrations of 20 µM (green peaks), 40 µM (purple peaks), and 200 µM (blue peaks). (C) The interaction of endogenous CXCL12 with i[VREY]4-biot (1 µM) in human blood was quantified on platelets by proximity ligation with DuoLink by flow cytometry (C) and visualized by confocal microscopy on platelets (scale bar, 2 µm) (n = 3) (D). Data represent mean ± standard deviation from the indicated numbers of independent experiments. **P ≤ .01, ***P ≤ .001 as analyzed by one-way analysis of variance with Dunnett’s multiple comparison test (panel C). MFI, mean fluorescence intensity.
Affinity differences between surface plasmon resonance and NMR are likely due to different protein concentrations, pH, and/or conformational changes induced by surface binding. We found that i[VREY]4 binds to the surface of resting human platelets in complex with endogenous CXCL12, as shown by antibody-based proximity ligation analyzed by using flow cytometry and visualized with fluorescence microscopy (Figure 3C-D). In addition, we observed that activation of platelets with collagen promoted the presentation of CXCL12 and increased i[VREY]4 binding and their complex formation on the surface of human platelets (supplemental Figure 4).
Functionally, i[VREY]4 inhibited platelet aggregation in human blood induced by low-dose collagen in combination with CXCL12 or by CXCL12 and collagen as single agonists (Figure 4A-C). The inhibitory effect of i[VREY]4 on collagen-induced platelet aggregation could be explained by a secondary release of CXCL12. Likewise, platelet aggregation induced by human plaque homogenate was inhibited by i[VREY]4 (Figure 4D). In a plaque-coated flow chamber perfused with human blood, i[VREY]4 decreased thrombus volume ex vivo (Figure 4E). Upon FeCl3 application, i[VREY]4 injected intraperitoneally effectively reduced arterial thrombosis in vivo (Figure 4F). To test whether the activity of i[VREY]4 requires platelet-derived CXCL12, we compared collagen-induced platelet aggregation in blood from Cxcl12Δplt/Δplt mice and Cxcl12wtlwt mice. In blood collected from Cxcl12Δplt/Δplt mice, collagen activation resulted in lower platelet aggregation than in that from Cxcl12wtlwt mice (Figure 4G). i[VREY]4 diminished platelet aggregation in blood from Cxcl12wtlwt mice but not from Cxcl12Δplt/Δplt mice. As negative controls, CCL1 and VREY did not affect platelet aggregation, and VREY did not inhibit thrombus formation ex vivo or in vivo (supplemental Figure 5; supplemental Videos 1-3). These data indicate that the inhibitory effect of i[VREY]4 depends on platelet-derived CXCL12.
![i[VREY]4 inhibits the prothrombotic activity of CXCL12. (A-D) The effects of i[VREY]4 (5 µM) on platelet aggregation in human blood activated with collagen and recombinant CXCL12 (0.1 µg/mL; n = 15) (A), recombinant CXCL12 alone (0.1 µg/mL) (B), collagen alone (0.2 µg/mL; n = 5) (C), or homogenized human plaque (833 µg/mL, n = 4) (D) were measured by using MEA. (E) Thrombus formation was induced by perfusion of human blood through a plaque-coated flow chamber at 600 s−1. Thrombus volume in the absence and presence of i[VREY]4 (5 µM) was analyzed by confocal microscopy (n = 6-7). (F) Time to occlusion as in Figure 1A. i[VREY]4 (100 µg, n = 10) or saline control (n = 9) was injected intraperitoneally 1 hour before induction of thrombosis. (G) Mouse blood from the indicated genotypes was mixed with 1 µg/mL collagen in the presence or absence of 5 µM i[VREY]4, and platelet aggregation was measured by using MEA (n = 6-8). (H) i[VREY]4-biot plasma levels were detected at various time points after intraperitoneal (i.p.) injection of 75 µg by enzyme-linked immunosorbent assay. (I) Neutrophil mobilization from the bone marrow of C57BL/6 mice was assessed 1 hour and 2 hours after i.p. injection of PBS with 100 μg i[VREY]4 or 100 μg AMD3465 by using an automated blood counter (n = 3-7). Data represent mean ± standard deviation from the indicated numbers of independent experiments or mice. *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001 as analyzed by using one-way analysis of variance with Tukey’s multiple comparison test (panels A, G, and I), unpaired t test (panels B and E), paired t test (panels C and D), and Mann-Whitney U test (panel F). AU, arbitrary units; ns, not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/17/10.1182_blood.2020010140/4/m_bloodbld2020010140f4.png?Expires=1765884507&Signature=RDkeINXAYxwBjoqD6Hy7p-fOkKQIQgtnFcGvCW4ASCyQLux~0ptuBqT2BVOnvEYr74uuboUMzHx1IXA0yZBzIyYHeVBDsyhyAMzZAW4TueUu9K0t9VOy-JGvpTj29cfBqT9XHhmGgwGBtgutetdES11E9-1cI~I5V9gUH1QG-hDs0KMrtKLC9ylFh7ufc0v74g8vvKYc9AMYmbs8qygQ6kS~5a~P5sgYGoQhgKcUdxBQm~PQP0F4w8WSkbWnl11lXAmt8O-TuFctbf4tlZtR9rA5ZPV9zUyim3oGTn--enOEvdOWcq5zVqd5jugM~wXERO9wajvZ~jSGakxMOCv9BA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
i[VREY]4 inhibits the prothrombotic activity of CXCL12. (A-D) The effects of i[VREY]4 (5 µM) on platelet aggregation in human blood activated with collagen and recombinant CXCL12 (0.1 µg/mL; n = 15) (A), recombinant CXCL12 alone (0.1 µg/mL) (B), collagen alone (0.2 µg/mL; n = 5) (C), or homogenized human plaque (833 µg/mL, n = 4) (D) were measured by using MEA. (E) Thrombus formation was induced by perfusion of human blood through a plaque-coated flow chamber at 600 s−1. Thrombus volume in the absence and presence of i[VREY]4 (5 µM) was analyzed by confocal microscopy (n = 6-7). (F) Time to occlusion as in Figure 1A. i[VREY]4 (100 µg, n = 10) or saline control (n = 9) was injected intraperitoneally 1 hour before induction of thrombosis. (G) Mouse blood from the indicated genotypes was mixed with 1 µg/mL collagen in the presence or absence of 5 µM i[VREY]4, and platelet aggregation was measured by using MEA (n = 6-8). (H) i[VREY]4-biot plasma levels were detected at various time points after intraperitoneal (i.p.) injection of 75 µg by enzyme-linked immunosorbent assay. (I) Neutrophil mobilization from the bone marrow of C57BL/6 mice was assessed 1 hour and 2 hours after i.p. injection of PBS with 100 μg i[VREY]4 or 100 μg AMD3465 by using an automated blood counter (n = 3-7). Data represent mean ± standard deviation from the indicated numbers of independent experiments or mice. *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001 as analyzed by using one-way analysis of variance with Tukey’s multiple comparison test (panels A, G, and I), unpaired t test (panels B and E), paired t test (panels C and D), and Mann-Whitney U test (panel F). AU, arbitrary units; ns, not significant.
i[VREY]4 inhibits the prothrombotic activity of CXCL12. (A-D) The effects of i[VREY]4 (5 µM) on platelet aggregation in human blood activated with collagen and recombinant CXCL12 (0.1 µg/mL; n = 15) (A), recombinant CXCL12 alone (0.1 µg/mL) (B), collagen alone (0.2 µg/mL; n = 5) (C), or homogenized human plaque (833 µg/mL, n = 4) (D) were measured by using MEA. (E) Thrombus formation was induced by perfusion of human blood through a plaque-coated flow chamber at 600 s−1. Thrombus volume in the absence and presence of i[VREY]4 (5 µM) was analyzed by confocal microscopy (n = 6-7). (F) Time to occlusion as in Figure 1A. i[VREY]4 (100 µg, n = 10) or saline control (n = 9) was injected intraperitoneally 1 hour before induction of thrombosis. (G) Mouse blood from the indicated genotypes was mixed with 1 µg/mL collagen in the presence or absence of 5 µM i[VREY]4, and platelet aggregation was measured by using MEA (n = 6-8). (H) i[VREY]4-biot plasma levels were detected at various time points after intraperitoneal (i.p.) injection of 75 µg by enzyme-linked immunosorbent assay. (I) Neutrophil mobilization from the bone marrow of C57BL/6 mice was assessed 1 hour and 2 hours after i.p. injection of PBS with 100 μg i[VREY]4 or 100 μg AMD3465 by using an automated blood counter (n = 3-7). Data represent mean ± standard deviation from the indicated numbers of independent experiments or mice. *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001 as analyzed by using one-way analysis of variance with Tukey’s multiple comparison test (panels A, G, and I), unpaired t test (panels B and E), paired t test (panels C and D), and Mann-Whitney U test (panel F). AU, arbitrary units; ns, not significant.
In a translational approach, we analyzed the pharmacokinetic variables of i[VREY]4 and their effect on bone marrow leukocyte release compared with AMD3465 (Figure 4H-I).35 Plasma concentrations of i[VREY]4-biot were measured by using a sandwich enzyme-linked immunosorbent assay with streptavidin to capture i[VREY]4-biot and a monoclonal antibody to the C-terminus of CCL5 that recognizes i[VREY]4. We found that intraperitoneal injection of 75 µg i[VREY]4 peaked at a maximal plasma concentration of 1.97 µg/mL after 30 minutes and declined to 0.07 µg/mL after 120 minutes (Figure 4H). In contrast to the classical CXCR4 agonist AMD3465, i[VREY]4 did not lead to significant mobilization of leukocytes from the bone marrow 1 or 2 hours’ postinjection (Figure 4I).
CXCL12 signals via Btk and PI3Kβ
Low-dose collagen elicits platelet activation via its receptor GPVI by signaling through Btk.36 This can be abolished by Btk inhibitors.24 Stimulation of chronic lymphatic leukemia cells with CXCL12 results in CXCR4 signaling through Btk.37 However, involvement of Btk in platelet CXCR4 signaling has not yet been investigated. Here, we found that human platelets pretreated with the highly selective covalent Btk inhibitor remibrutinib38 did not aggregate in blood or platelet-rich plasma (PRP) after stimulation with CXCL12 alone or in combination with low-dose collagen (Figure 5A-B; supplemental Figure 6A-D). Furthermore, CXCL12, CRP-XL, and collagen stimulated tyrosine phosphorylation of Btk at positions Y223 and Y551 using either platelets in blood (Figure 5C) or in PRP (Figure 5D-F). Remibrutinib inhibited both Btk-Y551 phosphorylation (Figure 5D-E) and platelet aggregation measured in the same PRP samples.

CXCL12-dependent platelet aggregation requires signaling through Btk. (A-B) Blood was pretreated for 30 minutes at 37°C with dimethyl sulfoxide (DMSO) (0.1% solvent control) or remibrutinib (0.1 µM) for Btk inhibition. Platelet aggregation was assessed by MEA after activation with collagen (0.1 µg/mL) and recombinant CXCL12 (0.1 µg/mL) or recombinant CXCL12 alone (1 µg/mL). (C) Phosphorylation of Btk (pBTK) in human platelets treated with CXCL12 (1 µg/mL) was analyzed by flow cytometry (n = 3). (D-F) PRP prepared from human blood was preincubated with DMSO (0.1%, solvent control) or remibrutinib (1 µM) for 30 minutes at 37°C before stimulation with CXCL12 (D), 2.5 µg/mL CRP-XL (E), or CXCL12 and collagen (n = 3) (F). Platelet aggregation was stopped after 1, 2, or 5 minutes by CGS buffer, and representative western blots patterns (upper panels D of and E) and quantification of Btk Y551 phosphorylation compared with total Btk (lower panels) are shown. (F) Phosphorylation of Y223 per total Btk after stimulation with CXCL12 (0.1-10 µg/mL) is shown in a representative immunoblot and densitometric quantification (lower panel) (n = 3). (G-H) Platelet activation was assessed by PAC-1 (activated αIIbβ3) and P-selectin antibody staining with and without Btk inhibition (0.1 µM remibrutinib) before stimulation with indicated combinations of recombinant CXCL12 (0.1 μg/mL) and CRP-XL (0.01 μg/mL). The samples were analyzed by using flow cytometry (n = 6). Platelet aggregation was assessed by MEA after activation with collagen (0.1 µg/mL) and CXCL12 (0.1 µg/mL) or CXCL12 alone (1 µg/mL). Data are represented as mean ± SD. Significant differences between different treatment groups are marked with asterisks *P ≤ .05, **P ≤ .01, ***P ≤ 0.001, ****P ≤ .0001, while differences between time points (D-E) within the same group are marked with hashtags ####P ≤ .0001 as analyzed by paired (A-B) or unpaired (C), t test and two-way analysis of variance (ANOVA) with Dunnett’s multiple comparison test (D,E,G,H). *DMSO + CRP-XL vs remibrutinib + CRP-XL of each time point and # at each time point vs time point 0. AU, arbitrary units; MFI, mean fluorescence intensity; ns, not significant.
CXCL12-dependent platelet aggregation requires signaling through Btk. (A-B) Blood was pretreated for 30 minutes at 37°C with dimethyl sulfoxide (DMSO) (0.1% solvent control) or remibrutinib (0.1 µM) for Btk inhibition. Platelet aggregation was assessed by MEA after activation with collagen (0.1 µg/mL) and recombinant CXCL12 (0.1 µg/mL) or recombinant CXCL12 alone (1 µg/mL). (C) Phosphorylation of Btk (pBTK) in human platelets treated with CXCL12 (1 µg/mL) was analyzed by flow cytometry (n = 3). (D-F) PRP prepared from human blood was preincubated with DMSO (0.1%, solvent control) or remibrutinib (1 µM) for 30 minutes at 37°C before stimulation with CXCL12 (D), 2.5 µg/mL CRP-XL (E), or CXCL12 and collagen (n = 3) (F). Platelet aggregation was stopped after 1, 2, or 5 minutes by CGS buffer, and representative western blots patterns (upper panels D of and E) and quantification of Btk Y551 phosphorylation compared with total Btk (lower panels) are shown. (F) Phosphorylation of Y223 per total Btk after stimulation with CXCL12 (0.1-10 µg/mL) is shown in a representative immunoblot and densitometric quantification (lower panel) (n = 3). (G-H) Platelet activation was assessed by PAC-1 (activated αIIbβ3) and P-selectin antibody staining with and without Btk inhibition (0.1 µM remibrutinib) before stimulation with indicated combinations of recombinant CXCL12 (0.1 μg/mL) and CRP-XL (0.01 μg/mL). The samples were analyzed by using flow cytometry (n = 6). Platelet aggregation was assessed by MEA after activation with collagen (0.1 µg/mL) and CXCL12 (0.1 µg/mL) or CXCL12 alone (1 µg/mL). Data are represented as mean ± SD. Significant differences between different treatment groups are marked with asterisks *P ≤ .05, **P ≤ .01, ***P ≤ 0.001, ****P ≤ .0001, while differences between time points (D-E) within the same group are marked with hashtags ####P ≤ .0001 as analyzed by paired (A-B) or unpaired (C), t test and two-way analysis of variance (ANOVA) with Dunnett’s multiple comparison test (D,E,G,H). *DMSO + CRP-XL vs remibrutinib + CRP-XL of each time point and # at each time point vs time point 0. AU, arbitrary units; MFI, mean fluorescence intensity; ns, not significant.
The activation of platelets in human blood by CRP-XL combined with CXCL12 increased P-selectin expression and integrin αIIbβ3 activation compared with each agonist alone (Figure 5G-H). This action was reversed by remibrutinib, indicating that both P-selectin and αIIbβ3 activation by GPVI and CXCR4 require Btk signaling. In platelets, Btk can be activated by Syk-mediated phosphorylation at Y223 and by binding to PIP3 generated via PI3K, which is part of the CXCL12-signaling pathway and a central component activating Btk.3,8 We observed that the Syk inhibitor II completely prevented collagen-induced platelet aggregation and strongly reduced the aggregation induced by CXCL12 and its combination with collagen (supplemental Figure 6E). Using the PI3K inhibitor TGX-221 specific for the p110β isoform, we observed that blocking PI3Kβ abolished platelet aggregation by CXCL12 alone and strongly inhibited aggregation induced in combination with collagen (supplemental Figure 6F-G). Targeting further events of the CXCL12 signaling cascade,9 we found that inhibition of p38 MAP kinase by SB2035080 and intracellular calcium release almost fully blocked platelet aggregation induced by CXCL12 alone and reduced that induced by collagen and CXCL12 in combination (supplemental Figure 6H-J). We conclude that platelet activation by CXCL12 requires similar signaling components as low-dose collagen.
i[VREY]4 binds CXCL12 to inhibit Btk activation but not CXCR4 binding
Incubation of blood with CXCL12 alone resulted in Btk phosphorylation, a process that could be inhibited by pretreatment with i[VREY]4 (Figure 6A), suggesting a CXCL12-dependent mechanism of i[VREY]4. In contrast, internalization of CXCR4 after CXCL12 exposure could not be reversed by i[VREY]4 (Figure 6B), consistent with biased signaling. Both exogenous CXCL12 and endogenous CXCL12 released by collagen treatment could be detected by a nonblocking antibody (clone #79018) or by a blocking antibody (K15C) directed to the N-terminal region of CXCL12, an interaction that occurs only with protomeric or GAG-bound CXCL12 that is not associated with CXCR4. Addition of i[VREY]4 did not result in reduced binding of K15C (Figure 6C-D), suggesting that i[VREY]4 does not affect binding of CXCL12 to GAGs on the platelet surface. In contrast, binding of clone #79018 was diminished by i[VREY]4 (Figure 6E-F), indicating that binding of i[VREY]4 to CXCL12 does not require CXCL12 motifs bound to CXCR4. To directly assess whether i[VREY]4 binding is influenced by the presence of CXCR4, we compared the binding of i[VREY]4-biot vs resting and collagen-stimulated platelets from wild-type and Cxcr4-deficient mice (Figure 6G). Similar to results with human platelets (supplemental Figure 4B), the robust binding of i[VREY]4 to the surface of mouse platelets required platelet activation and CXCR4. No binding was observed to the surface of unactivated platelets, making a direct interaction with CXCR4 unlikely. These data are consistent with a ternary complex formed between i[VREY]4, CXCL12, and CXCR4 on the platelet surface.
![i[VREY]4 blocks CXCL12-induced phosphorylation of Btk (pBTK) CXCR4 dependently without affecting CXCR4 internalization. (A) pBTK in human platelets was analyzed by using flow cytometry. Platelets were treated with CXCL12 (1 µg/mL) and as indicated with i[VREY]4 (n = 3). (B) Changes in CXCR4 expression on human platelets was analyzed by flow cytometry after treatment with recombinant CXCL12 (0.1 µg/mL), collagen (1 µg/mL), and i[VREY]4 (5 µM). (C-F) CXCL12 on human platelets was detected by flow cytometry. Human blood was treated with CXCL12 (0.1 µg/mL) (C,E) or collagen (1 µg/mL) (D,F), and detection was conducted with directly conjugated monoclonal antibodies (panels C and D, clone K15C, n = 10; panels E and F, clone 79018, n = 8). Binding of i[VREY]4-biot to platelets from tamoxifen-injected CreErtwt/wt Cxcr4flox/flox (wild type [WT]) or CreErttg/wt Cxcr4flox/flox (CXCR4 knockout [KO]) mice was measured by flow cytometry under resting conditions or stimulated with 10 µg/mL collagen (G) (n = 3-5). Data represent mean ± standard deviation from the indicated numbers of independent experiments or mice. *P ≤ .05, **P ≤ .01, as analyzed by one-way analysis of variance with Tukey’s multiple comparison test (panels A-F) or unpaired t test (panel G). MFI, mean fluorescence intensity; ns, not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/17/10.1182_blood.2020010140/4/m_bloodbld2020010140f6.png?Expires=1765884507&Signature=CggmV34WdXdzIlgjAvbLuuTIAvg3DofYwx7BHrbKR7aUYwqEviW6vKd8UF4Mw~-CX0P0je~zaIrcd8PVTkYakQLdcAToujElwezhKX3V7JUnECtUAwdFQqlzmKUAhsMMIFttT1fGUdwaPP0OdCjIx9WmPoi2lOrKwZZK5ogj1kMuIZ7eAdLYLXnY-FKtoGA2~8Rt3TPIJyoVJ~RPrVeveGyHT1OqPzsIreauxZ6GeuryAqD~fdkqCXBLbhByTE6Owpt0MmXTJCYJloHN1Zdk-2knZRiRIWSjB~Z2kcfXgyh6x3ZzyXs-NVU5FOQYZI2SdxSLodPCO3qHBhQdtK~BYQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
i[VREY]4 blocks CXCL12-induced phosphorylation of Btk (pBTK) CXCR4 dependently without affecting CXCR4 internalization. (A) pBTK in human platelets was analyzed by using flow cytometry. Platelets were treated with CXCL12 (1 µg/mL) and as indicated with i[VREY]4 (n = 3). (B) Changes in CXCR4 expression on human platelets was analyzed by flow cytometry after treatment with recombinant CXCL12 (0.1 µg/mL), collagen (1 µg/mL), and i[VREY]4 (5 µM). (C-F) CXCL12 on human platelets was detected by flow cytometry. Human blood was treated with CXCL12 (0.1 µg/mL) (C,E) or collagen (1 µg/mL) (D,F), and detection was conducted with directly conjugated monoclonal antibodies (panels C and D, clone K15C, n = 10; panels E and F, clone 79018, n = 8). Binding of i[VREY]4-biot to platelets from tamoxifen-injected CreErtwt/wt Cxcr4flox/flox (wild type [WT]) or CreErttg/wt Cxcr4flox/flox (CXCR4 knockout [KO]) mice was measured by flow cytometry under resting conditions or stimulated with 10 µg/mL collagen (G) (n = 3-5). Data represent mean ± standard deviation from the indicated numbers of independent experiments or mice. *P ≤ .05, **P ≤ .01, as analyzed by one-way analysis of variance with Tukey’s multiple comparison test (panels A-F) or unpaired t test (panel G). MFI, mean fluorescence intensity; ns, not significant.
i[VREY]4 blocks CXCL12-induced phosphorylation of Btk (pBTK) CXCR4 dependently without affecting CXCR4 internalization. (A) pBTK in human platelets was analyzed by using flow cytometry. Platelets were treated with CXCL12 (1 µg/mL) and as indicated with i[VREY]4 (n = 3). (B) Changes in CXCR4 expression on human platelets was analyzed by flow cytometry after treatment with recombinant CXCL12 (0.1 µg/mL), collagen (1 µg/mL), and i[VREY]4 (5 µM). (C-F) CXCL12 on human platelets was detected by flow cytometry. Human blood was treated with CXCL12 (0.1 µg/mL) (C,E) or collagen (1 µg/mL) (D,F), and detection was conducted with directly conjugated monoclonal antibodies (panels C and D, clone K15C, n = 10; panels E and F, clone 79018, n = 8). Binding of i[VREY]4-biot to platelets from tamoxifen-injected CreErtwt/wt Cxcr4flox/flox (wild type [WT]) or CreErttg/wt Cxcr4flox/flox (CXCR4 knockout [KO]) mice was measured by flow cytometry under resting conditions or stimulated with 10 µg/mL collagen (G) (n = 3-5). Data represent mean ± standard deviation from the indicated numbers of independent experiments or mice. *P ≤ .05, **P ≤ .01, as analyzed by one-way analysis of variance with Tukey’s multiple comparison test (panels A-F) or unpaired t test (panel G). MFI, mean fluorescence intensity; ns, not significant.
i[VREY]4 improves effects of standard antiplatelet therapy without affecting bleeding
To compare the effects of i[VREY]4 with those of standard antiplatelet therapies and to test whether a combination would offer added benefit, we assessed platelet aggregation by MEA and in vitro bleeding time using the platelet function analyzer–100/200 device, which is highly sensitive to conditions that affect primary hemostasis.39 Incubating human blood with aspirin resulted in prolonged closure time that exceeded the limit of 300 seconds, whereas the direct P2Y12 antagonist cangrelor prolonged closure time to a lesser extent. Incubation of blood with i[VREY]4 did not cause prolongation of the closure time beyond normal values (<120 seconds) or increase the closure time of cangrelor (Figure 7A). Comparing the effects on platelet aggregation induced by CXCL12 alone or combined with collagen, we observed that cangrelor showed only a small inhibitory tendency that could be enhanced by adding i[VREY]4 (Figure 7B-C).
![i[VREY]4 improves the inhibitory effect of standard antiplatelet therapy without increasing the risk of bleeding. (A) The effect of aspirin (300 µg/mL), cangrelor (0.34 µg/mL), and i[VREY]4 (5 µM) alone or in combination on collagen/epinephrine closure time was measured with the platelet function analyzer-200 device (n = 5). (B-G) Platelet aggregation was assessed by MEA in human blood activated with collagen (0.1 µg/mL) and recombinant CXCL12 (0.1 µg/mL), CXCL12 alone (1 µg/mL), or human plaque homogenate (833 µg/mL). The blood was pretreated for 1 hour either with dimethyl sulfoxide as a control, aspirin (300 µg/mL) alone, or in combination with i[VREY]4 (5 µM) or cangrelor (0.34 µg/mL) alone or in combination with i[VREY]4 (n = 8). Data represent mean ± standard deviation from the indicated numbers of independent experiments. *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001 as analyzed by repeated measure one-way analysis of variance with Tukey’s multiple comparison test. AU, arbitrary units; ns, not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/17/10.1182_blood.2020010140/4/m_bloodbld2020010140f7.png?Expires=1765884507&Signature=ZuD0T0LT4c2gH4FcWngKRjps~51151eXYmu6HWa7UG-ZNMcC7AjwsBeG9ol7lVqxc52Qi8VMCrnKANr8Oeyyl4cIbAUHAN7jbgw5uosRV2-bSJK1QN1ibY~VlDeGpl5nLTsYJV0TNjBXqYznwy3DOrR1rvDlwCqE0BOtmmz-6Na5843xQKjEBskx2mabCxRUI~334PVEMTqRVivVB45L-1a5hk~XI3OfL-gTqr3jJb6QASC789o4WwOM5ckKrpyNNKp8vP6oeDgutb4n9ntG6Emx1BIutz7b1EpQnfONFBlPAfc3anOyEWFHGwA0Jucnd1YWsW60~48yJ9xnfkF4qg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
i[VREY]4 improves the inhibitory effect of standard antiplatelet therapy without increasing the risk of bleeding. (A) The effect of aspirin (300 µg/mL), cangrelor (0.34 µg/mL), and i[VREY]4 (5 µM) alone or in combination on collagen/epinephrine closure time was measured with the platelet function analyzer-200 device (n = 5). (B-G) Platelet aggregation was assessed by MEA in human blood activated with collagen (0.1 µg/mL) and recombinant CXCL12 (0.1 µg/mL), CXCL12 alone (1 µg/mL), or human plaque homogenate (833 µg/mL). The blood was pretreated for 1 hour either with dimethyl sulfoxide as a control, aspirin (300 µg/mL) alone, or in combination with i[VREY]4 (5 µM) or cangrelor (0.34 µg/mL) alone or in combination with i[VREY]4 (n = 8). Data represent mean ± standard deviation from the indicated numbers of independent experiments. *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001 as analyzed by repeated measure one-way analysis of variance with Tukey’s multiple comparison test. AU, arbitrary units; ns, not significant.
i[VREY]4 improves the inhibitory effect of standard antiplatelet therapy without increasing the risk of bleeding. (A) The effect of aspirin (300 µg/mL), cangrelor (0.34 µg/mL), and i[VREY]4 (5 µM) alone or in combination on collagen/epinephrine closure time was measured with the platelet function analyzer-200 device (n = 5). (B-G) Platelet aggregation was assessed by MEA in human blood activated with collagen (0.1 µg/mL) and recombinant CXCL12 (0.1 µg/mL), CXCL12 alone (1 µg/mL), or human plaque homogenate (833 µg/mL). The blood was pretreated for 1 hour either with dimethyl sulfoxide as a control, aspirin (300 µg/mL) alone, or in combination with i[VREY]4 (5 µM) or cangrelor (0.34 µg/mL) alone or in combination with i[VREY]4 (n = 8). Data represent mean ± standard deviation from the indicated numbers of independent experiments. *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001 as analyzed by repeated measure one-way analysis of variance with Tukey’s multiple comparison test. AU, arbitrary units; ns, not significant.
Aspirin is known to block collagen-induced platelet aggregation measured by MEA.26 We found that aspirin blocked platelet aggregation induced by a combination of collagen and CXCL12 but not by CXCL12 alone (Figure 7D-E). CXCL12-dependent platelet aggregation seems to be independent of ADP and thromboxane formation. Both platelet aggregation induced by CXCL12 alone or combined with collagen in the presence of aspirin could be further diminished by i[VREY]4. Moreover, adding i[VREY]4 to cangrelor enhanced reduction of plaque-induced platelet aggregation, whereas adding i[VREY]4 to aspirin had no effect (Figure 7F-G).
Discussion
The current study showed that the contribution of CXCL12 to arterial thrombosis depends on platelets as a source of CXCL12 that by itself, or amplifying the effects of atherosclerotic plaque material (including collagen), drives thrombus size and stability. We found that CXCL12 transmits signals via CXCR4 that activate platelets through PI3Kβ, Syk, Btk, intracellular calcium release, and MAPK. Thrombus formation in vivo and CXCL12-induced platelet aggregation can be inhibited by the peptide antagonist i[VREY]4 that binds to CXCL12 on the platelet surface and prevents CXCR4 signaling. This provides an innovative pharmacologic concept that could complement standard antiplatelet therapy.
Using a thrombosis model of the common carotid artery, we found that mice specifically deficient in CXCL12 of the megakaryocyte–platelet lineage form occlusive thrombi to a lesser extent, and vessel occlusion is more unstable than in littermate controls. Readouts from a standardized multiparametric experiment, in which mouse blood is perfused through a collagen-coated flow chamber, indicate decreased stability and reduced αIIbβ3 activation as the underlying mechanism, as the initial surface deposition of platelets is only slightly different for both genotypes, whereas the size of the growing thrombus and its contraction score are much smaller in the knockout. After stimulation with collagen, platelets from knockout mice exhibit lower levels of P-selectin and αIIbβ3 activation compared with littermate controls, reflecting that secondary release of CXCL12 by collagen triggers α-granule release and αIIbβ3 activation. The CXCL12-dependent upregulation of P-selectin mirrors the secretory response of platelets and is likely not the main cause for the difference in thrombus formation, although thrombus stability may partially depend on P-selectin.40 In human blood, microscopy revealed that blocking CXCR4 prevents the three-dimensional growth of in vitro thrombus formation under flow conditions, suggesting that the same mechanisms apply to human thrombus formation. Platelet aggregation measured in blood by using the multiplate device is highly sensitive to activation by collagen,26 and CXCL12 alone was sufficient to dose dependently trigger platelet aggregation, which is enhanced by low-dose collagen.
We found that CXCL12 results in phosphorylation of Btk in platelets at Y223 and Y551, which can be prevented by remibrutinib,38 a covalent, highly selective irreversible Btk inhibitor. Indeed, Btk activation seemed to be a central signaling hub. Previous studies found that CXCR4 activation leads to downstream signaling via Btk in leukemia cells,37 but it remained unclear whether this mechanism applies to other cell types such as platelets.
Btk is a known downstream target of the primary collagen receptor GPVI. Our results show for the first time that Btk in platelets is also activated by CXCL12 stimulation of CXCR4, a G protein–coupled receptor (GPCR). This is remarkable, as platelet activation by other stimuli of GPCRs (eg, thrombin receptor activating peptide, ADP, thromboxane) does not require Btk signaling.24,41 In future studies, it would be interesting to elucidate the signaling cascade downstream of platelet CXCR4 that leads to the activation of Btk.
P-selectin plays an important role in atherogenesis and neointimal hyperplasia via the formation of platelet–leukocyte complexes and deposition of platelet–chemokines.42,43 After vascular injury, local CXCL12 and CXCR4 contribute to neointimal hyperplasia through the recruitment of bone marrow–derived smooth muscle cells.44 Accordingly, neointima formation tended to be smaller in Cxcl12Δplt/Δplt mice, whereas atherosclerotic plaques did not differ. We explain this discrepancy by the fact that platelet CXCL12 is not directly involved in early atherosclerosis but rather subsequently via the size, structural quality, and molecular composition of the thrombus. Although this is not the core focus of this study, these results warrant further investigation to dissect the contribution of thrombosis and local mediators for neointima formation.
Previously, we synthesized [VREY]4, a TASP-01–scaffolded peptide consisting of 4 peptides derived from the CCL5 C-terminal helix (VREY) that inhibits CXCL12-induced platelet activation.20 Here, we report on the improved variant i[VREY4]4 that differs in its scaffold (TASP-02) and exhibits improved stability. Interaction studies of CXCL12 binding to i[VREY]4 or CCL5 unravel a much higher (100-fold) binding affinity between CXCL12 and i[VREY]4 (KD, 5.6 ± 0.6 nM) than that to CCL5 (KD, 578 ± 61 nM).20 When incubated with human or mouse blood, i[VREY]4 blocks platelet activation and aggregation induced by CXCL12 alone or in combination with low-dose collagen or by using homogenized human plaque material. Platelet activation by collagen and plaque homogenate results in the release of platelet chemokines, including CCL5, which has been shown to inhibit CXCL12-induced platelet activation.18,45 In platelets, CXCL12 and CCL5 are expressed at similar copy numbers but may be released with distinct kinetics, implying endogenous regulatory mechanisms, which render the point of interference and mode of action proposed for i[VREY]4 highly plausible.14,34,46,47 The difference in affinities for CXCL12 and the multivalent binding exhibited by i[VREY]4, however, might explain why i[VREY]4 is superior to endogenous CCL5 in inhibiting CXCL12.
Addition of i[VREY]4 to the P2Y12 inhibitor cangrelor or to aspirin further reduces platelet aggregation induced by CXCL12 and collagen. In terms of primary hemostasis and bleeding, i[VREY]4 may be advantageous, as deletion of platelet-derived CXCL12 does not prolong tail bleeding time, and i[VREY]4, unlike aspirin and cangrelor, did not increase closure time on the collagen/epinephrine cartridges of the platelet function analyzer-100 device. From a pharmacodynamic perspective, i[VREY]4 could thus be a suitable substitute or adjunct for established antiplatelet therapies. In our experimental setup, i[VREY]4 was effective when given 1 hour before thrombosis, reaching its maximum plasma levels during this time and then dissipating rapidly. Therefore, i[VREY]4 could be applicable in acute myocardial infarction or stroke. Because the antithrombotic mechanism of i[VREY]4 is exerted by binding to CXCL12 on the platelet surface, a longer duration of action is conceivable but must still be experimentally verified.
CXCR4 or CXCL12 antagonists are currently in clinical use or have entered clinical trials, namely plerixafor (AMD3100) or the Spiegelmer NOX-A12, and may exert similar effects in inhibiting CXCL12-induced platelet activation. However, due to their action in the bone marrow, their use leads to mobilization of leukocytes into the circulation, an effect that is desirable for obtaining hematopoietic stem cells but is considered problematic for the treatment of thrombosis or for cardiovascular prevention.48 In this regard, i[VREY]4 behaves favorably, because with this construct, no leukocyte mobilization was observed. This merits further clarification but could be due to i[VREY]4 being scavenged by platelets, before it reaches the bone marrow, or to its distinct inhibitory mechanism for CXCR4.
Based on our findings, i[VREY]4 appears to bind to the surface of activated platelets using CXCL12 bound to CXCR4. This prevents an important part of CXCL12-induced signaling, namely Btk activation, whereas pathways required for CXCR4 internalization remain unaffected. With improved understanding of the complex signaling behavior of GPCRs that are subject to biased signaling and our current findings, it is conceivable that i[VREY]4 forms a ternary complex with CXCL12 and CXCR4 to exert its inhibitory effects. In this model, CXCL12 would not bind to CXCR4 in its native state but rather in an altered conformation that possibly prevents only some of the activation signals such as arrestin-mediated signals required for proper GPCR trafficking. A similar phenomenon was described for a peptide of the transmembrane region of CXCR4, which turned out to be a biased antagonist inhibiting G protein signaling but not arrestin-mediated receptor internalization.49 Homodimerization of CXCL12 entails biased agonism for CXCR4, such that dimeric CXCL12 fails to promote chemotaxis and even operates as a competitive inhibitor.50 Interaction analysis with CXCL12 and i[VREY]4 using NMR spectroscopy indicates that i[VREY]4 might indeed interact with CXCL12 in a similar fashion.
Our proximity ligation study showing complex formation between i[VREY] and CXCL12 on the surface of platelets indicates that CXCL12 can transmit signals that lead to internalization of CXCR4 (arrestin) even in the presence of i[VREY], whereas other signals leading to Btk phosphorylation51 and platelet aggregation are blocked by i[VREY]4. In this regard, pharmacologic intervention with the CXCR4–CXCL12 axis could be preferable to drugs inhibiting Btk, as bleeding events occurring with some Btk inhibitors are not fully explained.52 Platelets are cellular mediators that maintain the balance between bleeding and thrombosis. The idea that it is possible to selectively shift this balance and generate a pharmaceutical agent that inhibits platelet aggregation but does not cause bleeding has received support by the existence of patients with X-linked agammaglobulinemia who lack functional Btk without increased bleeding risk and by the development of GPVI inhibitors such as Revacept (advanceCOR).33,53
In summary, we found that platelet-derived CXCL12 promotes arterial thrombosis by activating platelets through CXCR4, leading to Btk signaling and αIIbβ3-dependent thrombus growth and stability, whereas primary hemostasis was unaffected. Exploiting and translating inhibitory effects of heterodimerization between CCL5 and CXCL12, we show that the CCL5-derived peptide i[VREY]4 binds to CXCL12, thereby inhibiting CXCR4, Btk activation, and platelet aggregation, resulting in reduced thrombus formation. Our study has established i[VREY]4 as a novel promising candidate for further therapeutic development in atherothrombosis.
Acknowledgments
J.L. was supported by the DZHK (German Centre for Cardiovascular Research). This work was supported by the National Science Foundation (BIR-961477), the University of Minnesota Medical School and the Minnesota Medical Foundation (K.H.M.), and by the Deutsche Forschungsgemeinschaft, SFB1123, A2 (P.v.H.), A7 (C.S. and K.S.), Z1 (R.T.A.M.), and INST 409/150-1 FUGG (C.W. and R.T.A.M.). N.J.J. received funding from the European Union’s Horizon 2020 research and innovation program under Marie Sklodowska-Curie grant agreement No. 766118. At Maastricht University, C.W. is Van de Laar professor of atherosclerosis, and K.H.M. is Van de Laar professor of structural biology. K.H.M. is also grateful to the Ludwig-Maximillian-University Center for Advanced Study and the Alexander von Humboldt Foundation for support during his 2019 sabbatical stay at the Ludwig-Maximillian-University. P.Z. is supported by the European Union’s Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement TICARDIO No. 813409. The graphical abstract was created with BioRender.com.
Authorship
Contribution: C.W. and P.v.H. conceived the study; J.L. and P.v.H. wrote the original draft; J.L., S.M.A., X.B., R.D., H.I., M.A., N.J.J., K.J., R.T.A.M., and P.Z. acquired and analyzed the data; C.S., Y.D., and R.B. provided essential tools; J.L., C.S., J.D., Y.D., K.S., W.S., K.J., J.W.M.H., T.M.H., K.H.M., C.W., and P.v.H. interpreted results; and C.W. and P.v.H. supervised the project. All authors reviewed and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Philipp von Hundelshausen, Institute for Cardiovascular Prevention, Ludwig-Maximilians-Universität München, Pettenkoferstr. 9, 80336 Munich, Germany; e-mail: Philipp.von_Hundelshausen@med.uni-muenchen.de.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal