

Abstract
Hemochromatosis (HC) is a genetically heterogeneous disorder in which uncontrolled intestinal iron absorption may lead to progressive iron overload (IO) responsible for disabling and life-threatening complications such as arthritis, diabetes, heart failure, hepatic cirrhosis, and hepatocellular carcinoma. The recent advances in the knowledge of pathophysiology and molecular basis of iron metabolism have highlighted that HC is caused by mutations in at least 5 genes, resulting in insufficient hepcidin production or, rarely, resistance to hepcidin action. This has led to an HC classification based on different molecular subtypes, mainly reflecting successive gene discovery. This scheme was difficult to adopt in clinical practice and therefore needs revision. Here we present recommendations for unambiguous HC classification developed by a working group of the International Society for the Study of Iron in Biology and Medicine (BIOIRON Society), including both clinicians and basic scientists during a meeting in Heidelberg, Germany. We propose to deemphasize the use of the molecular subtype criteria in favor of a classification addressing both clinical issues and molecular complexity. Ferroportin disease (former type 4a) has been excluded because of its distinct phenotype. The novel classification aims to be of practical help whenever a detailed molecular characterization of HC is not readily available.
Historical perspective
It is commonly accepted that the term “hemochromatosis” was coined by the German pathologist von Recklinghausen in 1889. Of note, this was during the Versammlung Deutscher Naturforscher (meeting of German scientists) held in Heidelberg, like the BIOIRON Society (formerly IBIS) meeting in 2019, which led to the current report. Following the description of patients with “bronze diabetes and cirrhosis” by French physicians led by Armand Trousseau in the mid-1800s, von Recklinghausen hypothesized that something circulating in the blood (“hemo-”) was responsible for skin and organ damage and pigmentation (“-chromatosis”) (HC). Recognizing excess iron as the etiology of organ toxicity took several decades and was attributable to Joseph Sheldon in 1935, who was also the first to suggest the genetic origin of the metabolic defect.1 Overall, these pioneers' works clearly defined a clinical-pathological entity caused by progressive iron accumulation and characterized by multiorgan damage (mainly in the liver, pancreas, joints, heart, and endocrine glands), without signs of anemia (to the contrary, some patients show mildly increased Hb levels).2-5 In the 1950s, ferrokinetic studies revealed abnormally increased intestinal iron absorption as the key pathophysiological feature of HC,6 and repeated/frequent phlebotomies were established as the mainstay of treatment.7 In 1977, the seminal work by Marcel Simon and colleagues reported the tight linkage between the major histocompatibility complex (MHC) and the putative hemochromatosis gene on chromosome 6p, definitively demonstrating the genetic origin of the disease.8 This paved the way to the discovery, in 1996, of the “hemochromatosis gene” HFE (alias “high Fe,” official full name “homeostatic iron regulator”),9 which provided additional information about HC. Initially, it appeared that up to 95% of HC cases could be attributed to homozygosity for a single nucleotide change (845 G→A), causing the substitution of cysteine by tyrosine at amino acid 282 (p.Cys282Tyr or C282Y variant).10-13 A second HFE polymorphism, p.His63Asp, was detected, whose minor role became clearer later.14 The meaning of the p.Cys282Tyr/p.His63Asp compound heterozygosity is discussed in detail below. The high frequency of p.Cys282Tyr homozygosity in the original studies resulted from the inclusion of patients mostly of Northern European ancestry, a region where the variant had originated around 4000 BC.15,16 Indeed, the p.Cys282Tyr variant, frequent in certain geographical regions,17 is rare or even absent in large areas of the world, including Asian and African countries, as well as in Native Americans.18,19 Subsequent studies in Southern Europe, in the Mediterranean area, and in Brazil found that at least one-third of subjects with a defined HC phenotype were negative for p.Cys282Tyr at HFE genetic testing.20,21
Over time, it became evident that the genetic basis of HC was more heterogeneous than initially assumed, and several variants in other iron-controlling genes (collectively referred to as “non-HFE genes”) were progressively associated with the disorder. These include variants on genes coding for a second receptor for transferrin (TFR2),22-24 ferroportin (SLC40A1),25 hepcidin (HAMP),26,27 and hemojuvelin (HJV).28,29 In particular, the discovery of variants in the HAMP and HJV genes made it possible to define a severe early-onset (juvenile) form of HC, with early cardiac and endocrine impairments, as a molecularly distinct entity.
Hepcidin is the master regulator of iron homeostasis,30-33 and its identification has represented a considerable advance in the comprehension of the pathophysiological mechanisms underlying HC. For detailed reviews on hepcidin discovery, functions and regulation, readers are referred elsewhere.34-37 Briefly, hepcidin is a small peptide hormone produced by the liver that negatively controls circulating iron levels. Through interaction with ferroportin38-40 (its receptor and the only cellular iron exporter so far identified in humans), hepcidin inhibits the absorption of dietary iron in the duodenum and its release by spleen macrophages involved in recycling iron from senescent erythrocytes. Molecular defects causing hepcidin deficiency result in uncontrolled intestinal iron absorption, with progressive iron accumulation in tissues, ultimately leading to HC.1 In most cases, gene defects cause insufficient production of hepcidin, while rarely ferroportin resistance to hepcidin is observed. As illustrated in Figure 1, hepcidin regulation by iron is quite complex and involves numerous proteins41,42 whose alterations can compromise hormone synthesis or function. For this reason, identifying the molecular causes of HC is far from simple and requires a deep knowledge of its pathogenetic basis, which is still not completely clarified. In our view, the term “hemochromatosis” should be reserved for this unique clinical entity caused by genetic lesions that primarily affect the hepcidin-ferroportin system and not used to describe clinically distinct iron overload conditions with other causes.
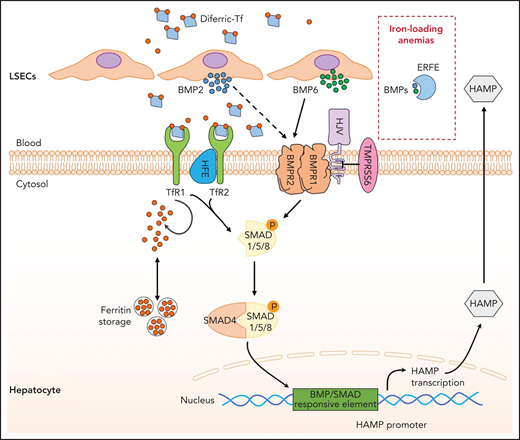
Hepcidin regulation by iron. Increase in transferrin saturation induces hepcidin transcription via the BMP/SMAD signaling pathway. Diferric transferrin binds to TfR2, while BMP6 and BMP2 secreted by liver sinusoidal endothelial cells (LSECs) bind to BMP receptors on hepatocytes. These events trigger phosphorylation of regulatory SMAD1/5/8, recruitment of SMAD4, and translocation of the SMAD complex to the nucleus for activating hepcidin transcription upon binding to BMP/SMAD responsive element in the HAMP promoter. BMPs can be trapped by ERFE, leading to hepcidin inhibition in iron-loading anemias. Efficient iron signaling requires the BMP coreceptor HJV and the protein HFE, and is negatively regulated by the transmembrane serine protease matriptase-2 (TMPRSS6). The complex molecular pathogenesis of HC reflects the numerous proteins involved in the regulation of the hepcidin-ferroportin axis.
Hepcidin regulation by iron. Increase in transferrin saturation induces hepcidin transcription via the BMP/SMAD signaling pathway. Diferric transferrin binds to TfR2, while BMP6 and BMP2 secreted by liver sinusoidal endothelial cells (LSECs) bind to BMP receptors on hepatocytes. These events trigger phosphorylation of regulatory SMAD1/5/8, recruitment of SMAD4, and translocation of the SMAD complex to the nucleus for activating hepcidin transcription upon binding to BMP/SMAD responsive element in the HAMP promoter. BMPs can be trapped by ERFE, leading to hepcidin inhibition in iron-loading anemias. Efficient iron signaling requires the BMP coreceptor HJV and the protein HFE, and is negatively regulated by the transmembrane serine protease matriptase-2 (TMPRSS6). The complex molecular pathogenesis of HC reflects the numerous proteins involved in the regulation of the hepcidin-ferroportin axis.
The current clinical scenario
Unlike in the past, fully expressed and potentially lethal HC (with liver cirrhosis, diabetes, endocrine dysfunction, and heart failure) is seen rarely in current clinical practice.43 This can be ascribed to increased awareness of the disease and, mostly, to the routine assessment of iron biomarkers, particularly serum ferritin. Unfortunately, this is counterbalanced by an increased diagnostic challenge for nonexperts in the iron field, primarily due to the lack of specificity of ferritin.
Ferritin is an essentially intracellular protein that serves to store iron safely. It is also present at very low concentrations (micrograms per liter) in serum, likely through secretion by macrophages.44 Normal values usually range from 30 to 200 µg/L or 300 µg/L in females and males, respectively. The function of secreted extracellular ferritin remains largely unknown.45,46 Several common conditions lead to increased serum ferritin levels, including virtually all inflammatory disorders, hepatic cytolysis (eg, during acute or chronic liver disease), or metabolic syndrome.47,48 This translates into a huge number of consultations, overuse of the “first-level” genetic test looking for the presence of the common variants in the HFE gene, and even misdiagnosis due to incorrect interpretation of the results.49
The glycoprotein transferrin is the extracellular carrier of iron that is detectable at high concentrations in blood (grams per liter; the third most abundant protein after albumin). Transferrin saturation (TSAT) is calculated as the ratio between serum iron and transferrin (multiplied by the correction factor 1.42) or, less reliably, between serum iron and total iron-binding capacity and expressed as a percentage. TSAT is much less requested in clinical practice but is much more informative about a possible diagnosis of HC. Normal TSAT varies between 20% and 45%. It has been estimated that hyperferritinemia with normal TSAT is associated with increased iron stores in less than 10% of cases.50 Importantly, TSAT elevation is the hallmark of HC. In HC patients, high TSAT reflects the increased pool of circulating iron due to insufficient hepcidin production and typically precedes the rise of serum ferritin by several years.51 TSAT tends to remain elevated even in subjects effectively iron-depleted by phlebotomies. Occasional reports of normal TSAT in p.Cys282Tyr homozygotes with hyperferritinemia should always prompt the search for additional cofactors that raise ferritin, such as metabolic syndrome or alcohol intake.52,53
Liver biopsy was once regarded as the gold standard for the diagnosis of HC because it can reveal iron deposition in hepatocytes with the typical decreasing gradient from the periportal zone (most exposed to iron coming from the gut) to the central-lobular zone in the hepatic acinus. However, in current practice, the demonstration of p.Cys282Tyr homozygosity along with elevated serum ferritin and TSAT is considered sufficient to make the diagnosis of HC. Liver biopsy remains useful for prognostic purposes in HC patients with serum ferritin levels repeatedly >1000 µg/L, allowing the early identification of advanced fibrosis or even subclinical cirrhosis. These conditions require close surveillance for hepatocellular carcinoma even after iron depletion.51 Nowadays, liver biopsy is seldom performed due to its invasiveness, costs, and the increasing availability of noninvasive tools. Indeed, magnetic resonance imaging (MRI) techniques have largely replaced it for the determination of liver iron concentration (LIC). This is obtained indirectly by using various MRI protocols, for which there is still no consensus on the best one. The choice of the protocol mainly depends on local expertise, as well as on the available equipment and software.54-56 Moreover, hepatic transient elastography (FibroScan) is a reliable noninvasive method for detecting liver fibrosis in HC patients, limiting the need for liver biopsy to those with indeterminate results.57
Currently, HC is typically suspected in subjects with no or minimal symptoms, increased serum ferritin levels without alternative explanation, high TSAT, and evidence of increased liver iron stores by MRI. Making the diagnosis at this preclinical early stage51 has the undoubted advantage of preventing organ damage by a relatively simple and cost-effective treatment (phlebotomy), as well as of allowing normal life expectancy.58,59
The HFE genetic test, available in most laboratories, identifies the most common inherited defect in Whites predisposing to HFE-related HC, ie, p.Cys282Tyr homozygosity. Therefore, in Whites, an HFE genetic test is indicated when high TSAT is confirmed irrespective of parenchymal iron overload (IO) demonstration. p.Cys282Tyr homozygosity has a variable and difficult to predict clinical penetrance,60-64 with several inherited and acquired modifiers potentially contributing to the final phenotype. For this reason, HFE-related HC should not be viewed as a simple monogenic disorder but rather as the complex result of the interplay of environmental, lifestyle, and still unidentified genetic cofactors.1
Regarding the compound p.Cys282Tyr and p.His63Asp heterozygosity, compelling evidence exists that this genotype per se is characterized by minimal or no clinical penetrance.65,66 Thus, it cannot be considered diagnostic for HC,14 but at most as a susceptibility factor that can be associated with mild-to-moderate IO only in case of digenic inheritance67 (see below) or when other predominant causes of liver disease are present, namely nonalcoholic fatty liver disease (NAFLD), alcohol, or hepatitis C virus (HCV). Of note, the latter 2 are known to cause acquired hepcidin suppression.41,68,69 According to existing guidelines, whenever a subject with p.Cys282Tyr/p.His63Asp compound heterozygosity has evidence of IO, a secondary cause of liver disease should be sought and treated,70,71 with phlebotomies possibly considered as an adjunctive treatment. On the other hand, the negative effects of an automatic HC (mis)diagnosis in p.Cys282Tyr/p.His63Asp compound heterozygotes are commonly seen at referral centers. They include patients’ and family members’ unnecessary anxiety, incomplete prior investigations (eg, serum ferritin but neither TSAT nor MRI), overlooking of other causes, and/or unnecessary treatment by phlebotomies.
Clinical elements that should raise a definite suspicion of HC are reported in Table 1. In Whites with a negative first-level HFE test (ie, p.Cys282Tyr homozygosity is not detected) and in non-Whites, a second-level genetic test should be considered in order to identify rarer variants in the HFE or in other genes known to be linked to hepcidin control. In general, these types of HC are less influenced by cofactors and characterized by a more severe and homogeneous clinical picture appearing at a younger age.72 Their molecular diagnosis is often complex since variants in HC genes other than HFE are typically private (ie, restricted to members of only 1 or a few families). To this end, modern approaches based on next-generation sequencing (NGS) have greatly expanded the diagnostic possibilities in rarer HC while at the same time opening enormous challenges of interpretation of the results. NGS is generally available only at referral centers and requires specific expertise to avoid misdiagnosis, with a long wait time for results (see below). Nonetheless, treatment of patients with a defined HC phenotype should not be delayed pending the result of the genetic test. Recently, NGS methods have also made it possible to estimate the global prevalence of HFE and non-HFE HC in different populations,19 as summarized in Table 2.
Main clinical, biochemical, and imaging elements for the suspicion of HC
| Leading |
|---|
| TSAT >45% (mainstay) |
| S-Ferritin >200 µg/L (females) or >300 µg/L (males) |
| Imaging evidence of liver IO (MRI* and/or biopsy†) |
| Iron deposits in hepatocytes (if biopsy is performed) |
| Absence of “predominant” acquired risk factors for hepcidin deficiency (eg, alcohol abuse or end-stage liver disease) and iatrogenic IO (eg, regular transfusions) |
| Absence of hematological signs of a primary RBC disorder, such as anemia‡ and/or reticulocytosis |
| Not always present |
| Signs and/or symptoms associated with IO: |
| • Skin pigmentation, asthenia |
| • Persistent increase of aminotransferases, hepatomegaly, cirrhosis, hepatocellular carcinoma |
| • Joint pain, arthritis, chondrocalcinosis, reduced bone mineral density |
| • Diabetes mellitus, hypopituitarism, hypoparathyroidism, hypogonadotropic hypogonadism |
| • Cardiomyopathy, heart failure, cardiac arrhythmias |
| Leading |
|---|
| TSAT >45% (mainstay) |
| S-Ferritin >200 µg/L (females) or >300 µg/L (males) |
| Imaging evidence of liver IO (MRI* and/or biopsy†) |
| Iron deposits in hepatocytes (if biopsy is performed) |
| Absence of “predominant” acquired risk factors for hepcidin deficiency (eg, alcohol abuse or end-stage liver disease) and iatrogenic IO (eg, regular transfusions) |
| Absence of hematological signs of a primary RBC disorder, such as anemia‡ and/or reticulocytosis |
| Not always present |
| Signs and/or symptoms associated with IO: |
| • Skin pigmentation, asthenia |
| • Persistent increase of aminotransferases, hepatomegaly, cirrhosis, hepatocellular carcinoma |
| • Joint pain, arthritis, chondrocalcinosis, reduced bone mineral density |
| • Diabetes mellitus, hypopituitarism, hypoparathyroidism, hypogonadotropic hypogonadism |
| • Cardiomyopathy, heart failure, cardiac arrhythmias |
TSAT, transferrin saturation; IO, iron overload; MRI, magnetic resonance imaging.
Liver iron concentration (LIC) by MRI can be obtained using different protocols, which vary depending on local expertise and equipment. With these limitations, any LIC value higher than the upper normal limit (generally set at 36 µmol/g to 40 µmol/g dry weight) should lead to consideration of phlebotomies in HC patients. Similarly, LIC >100 µmol/g to 120 µmol/g and >240 µmol/g to 300 µmol/g are generally considered as overt and severe IO, respectively (see text and references.55,56).
Liver biopsy should be considered in patients with ferritin >1000 µg/L or signs of liver damage.
Exceptions may occur (eg, in HC patients with diagnosis delayed after the appearance of liver cirrhosis, in whom anemia may be observed because of hypersplenism or gastrointestinal bleeding, or subjects with β-thalassemia trait, whose coexistence is not rare in Mediterranean countries).
Combined pathogenic allele frequency for HC genes in the 1000 Genomes Project (1000G), Exome Sequencing Project (ESP), and Exome Aggregation Consortium (ExAc) datasets
| Gene | 1000G | ESP6500 | ExAc | Geographical distribution |
|---|---|---|---|---|
| HFE (p.Cys282Tyr) | 0.013 | 0.048 | 0.0324 | Highest prevalence in Northern Europe |
| HFE (non-p.Cys282Tyr) | 0.001 | 0.0002 | 0.000307 | |
| HJV | 0.00074 | 0.000316 | Highest prevalence in Southern Asia | |
| TFR2 | 0.0004 | 0.0003 | 0.000102 | Most frequent among non-Finnish European populations |
| HAMP | 0.0002 | 0.0000165 | Several populations | |
| SLC40A1 | 0.0008 | 0.0009 | 0.00034 | Several populations (highest prevalence among Africans) |
| Gene | 1000G | ESP6500 | ExAc | Geographical distribution |
|---|---|---|---|---|
| HFE (p.Cys282Tyr) | 0.013 | 0.048 | 0.0324 | Highest prevalence in Northern Europe |
| HFE (non-p.Cys282Tyr) | 0.001 | 0.0002 | 0.000307 | |
| HJV | 0.00074 | 0.000316 | Highest prevalence in Southern Asia | |
| TFR2 | 0.0004 | 0.0003 | 0.000102 | Most frequent among non-Finnish European populations |
| HAMP | 0.0002 | 0.0000165 | Several populations | |
| SLC40A1 | 0.0008 | 0.0009 | 0.00034 | Several populations (highest prevalence among Africans) |
Modified from Wallace and Subramaniam.19
Figure 2 illustrates a possible algorithm for the diagnosis of HC, starting from clinical, biochemical, and imaging studies to molecular confirmation.
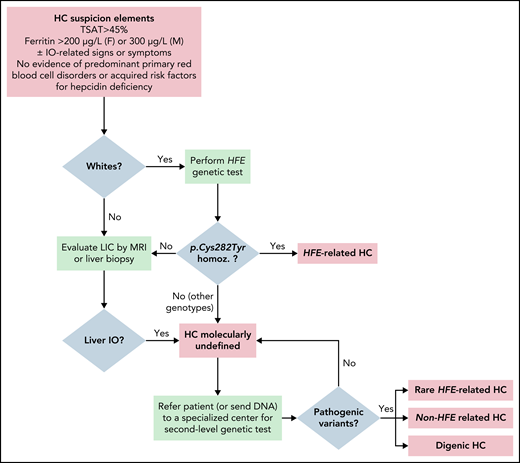
Proposal of an algorithm for the diagnosis of HC, from clinical/biochemical and imaging studies to molecular confirmation. Important note: in Whites, HFE genotyping is indicated with the specific purpose of detecting p.Cys282YTyr homozygosity (homoz.) and, if confirmed, to recommend appropriate preventive treatment by phlebotomies. Asian, African, and Native American subjects with defined HC phenotype could be directly referred to second-level genetic testing. In populations with a frequent component of Northern European ancestry, such as African Americans and Hispanics, there may still be a role for HFE genetic testing.
Proposal of an algorithm for the diagnosis of HC, from clinical/biochemical and imaging studies to molecular confirmation. Important note: in Whites, HFE genotyping is indicated with the specific purpose of detecting p.Cys282YTyr homozygosity (homoz.) and, if confirmed, to recommend appropriate preventive treatment by phlebotomies. Asian, African, and Native American subjects with defined HC phenotype could be directly referred to second-level genetic testing. In populations with a frequent component of Northern European ancestry, such as African Americans and Hispanics, there may still be a role for HFE genetic testing.
The nomenclature of genetic disorders
HC nomenclature suffers from a common problem of classifying genetic diseases. In contrast to genes, diseases lack a standardized way to review official names and symbols by formal committees (https://ghr.nlm.nih.gov/primer/mutationsanddisorders/naming). Disease nomenclature is often derived from the name(s) of the physician(s) who first described the condition, 1 major sign or symptom, or the biochemical/genetic underlying defect. However, the growing comprehension of the pathophysiological or molecular mechanisms that regulate diseases, as well as the identification of new phenotypes, may require revision of the initial name by experts in order to improve its usefulness in clinical practice.
Proper nomenclature, in fact, is an essential prerequisite for clear and effective communication about a particular condition. Ideally, it should unequivocally evoke disorders sharing the same pathogenesis and treatment, eventually helping clinicians to provide an accurate diagnosis and management.
What HC is (and what it is not)
Nomenclature and case definition of HC have long been recognized as potential sources of confusion,73 especially when dealing with the report of genetic tests.14 Among experts, there was a common feeling that the HC classification needs to be revised in view of the increasing awareness and knowledge of IO disorders. To this end, the BIOIRON Society promoted a 2-step process. The starting point was the preparation of a survey that was sent to working group participants, including both expert clinicians and basic scientists actively involved in the iron metabolism field, and consisting of nearly all who discovered the hemochromatosis genes and hepcidin. The survey questions and the summary of responses are available in the supplemental Material (available on the Blood Web site). This was followed by a critical collegial discussion during a specific session of the most recent biennial meeting of the BIOIRON Society in Heidelberg. The recommendations reported here are the result of such discussion, where the panelists eventually agreed on the novel classification. As an integral part of the process, the panelists agreed on a robust definition of HC, based on clinical presentation and widely available tools, as a prerequisite for genetic testing. The main clinical, biochemical, and imaging studies for the suspicion of HC are reported in Table 1. Rigorously speaking, the term “hemochromatosis” should be reserved for a unique genetic clinical-pathological condition characterized by increased TSAT, IO in the liver (but not in the spleen), prevalent involvement of periportal hepatocytes with iron-spared Kupffer cells, and signs and/or symptoms associated with IO. The panelists also emphasized that the term “hemochromatosis” itself implies an IO of genetic origin, which is why they would recommend avoiding the unnecessary use of qualifiers such as “hereditary,” “genetic,” or “primary.” Indeed, genetic defects in the hepcidin/ferroportin regulatory axis (caused by variants in hepcidin regulators, the hepcidin gene itself, or in ferroportin) are responsible for inadequate production or activity of hepcidin or lack of hepcidin responsiveness of ferroportin.
Finally, the panelists agreed that the definition of HC should also include the absence of hematological signs of a primary/predominant red blood cell (RBC) disorder, such as anemia or reticulocytosis (see Table 1 for some exceptions to this rule). This is needed to distinguish HC from other IO conditions, often referred to as “iron-loading anemias,”74,75 which are similarly characterized by increased TSAT and are nearly always genetically determined. In these conditions, the hepcidin suppression is caused by factors released by erythropoietin-stimulated erythroblasts (eg, erythroferrone or ERFE),76 as a consequence of ineffective erythropoiesis or compensated chronic hemolysis, and not by variants in genes affecting the hepcidin-ferroportin axis. The prototype of this group is nontransfusion dependent thalassemia (NTDT).77 IO also occurs in transfusion-dependent inherited anemias, but in this case, it is mainly due to transfusions per se, and hepcidin levels tend to be increased,78 especially immediately after the transfusion because of suppression of erythropoiesis.79Figure 3 illustrates the main mechanisms underlying the development of IO in hemochromatosis and iron loading anemias. As mentioned above, the majority of iron loading anemias are inherited,36,80-82 including forms caused by variants in the hemoglobin genes, in genes coding for RBC enzymes or membrane structures, as well as congenital sideroblastic81 or dyserythropoietic82 anemias. Sometimes, variants in genes directly regulating iron transport and utilization (such as DMT1, transferrin, ceruloplasmin, and others) may be implicated as well.80 Finally, IO due to hepcidin inhibition can also occur independently of transfusions in some forms of myelodysplastic syndromes,83 especially those characterized by ringed sideroblasts and increased ineffective erythropoiesis associated with acquired somatic mutations in SF3B1.84 In any case, all these conditions should never be regarded as hemochromatosis because of the distinct pathogenesis and treatment (eg, phlebotomies are often not feasible).
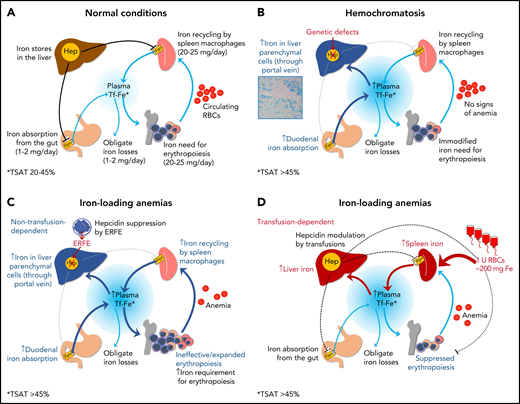
Iron homeostasis in normal conditions (A) and mechanisms leading to iron accumulation in HC (B) and in iron-loading anemias that are nontransfusion-dependent (C) and transfusion-dependent (D). In HC, iron hyperabsorption through the portal vein leads to iron accumulation in liver parenchymal cells, initially with a typical portal-central gradient (see histology) and sparing of macrophages (Kupffer cells). In nontransfusion-dependent anemias with ineffective erythropoiesis, hepcidin insufficiency is also central to the pathogenesis of IO, but it is due to suppression by soluble factors (eg, ERFE) produced by ineffective/expanded erythroblasts rather than to a genetic defect in pathways regulating hepcidin synthesis. In transfusion-dependent anemias, regular red blood cells (RBCs) transfusions represent the major contributing factor to IO; in these conditions, hepcidin is relatively upregulated by iron but fluctuates in response to intermittent erythropoiesis suppression by transfusions.
Iron homeostasis in normal conditions (A) and mechanisms leading to iron accumulation in HC (B) and in iron-loading anemias that are nontransfusion-dependent (C) and transfusion-dependent (D). In HC, iron hyperabsorption through the portal vein leads to iron accumulation in liver parenchymal cells, initially with a typical portal-central gradient (see histology) and sparing of macrophages (Kupffer cells). In nontransfusion-dependent anemias with ineffective erythropoiesis, hepcidin insufficiency is also central to the pathogenesis of IO, but it is due to suppression by soluble factors (eg, ERFE) produced by ineffective/expanded erythroblasts rather than to a genetic defect in pathways regulating hepcidin synthesis. In transfusion-dependent anemias, regular red blood cells (RBCs) transfusions represent the major contributing factor to IO; in these conditions, hepcidin is relatively upregulated by iron but fluctuates in response to intermittent erythropoiesis suppression by transfusions.
The former classifications: strengths and shortcomings
The HC classifications reported by authoritative textbooks and recent reviews and guidelines are based on a schema (Table 3) in which numbers and letters reflect the chronology of first descriptions of genotype-phenotype correlations.51,71,85,86 Four types are included, with type 2 and type 4 further subdivided into subtypes A and B. They have the advantage of being very informative from a molecular point of view and officially endorsed by OMIM (the Online Mendelian Inheritance of Man database), but also present several caveats and inconsistencies. The main limitations of the current classifications are listed below.
Former classification of HC
| Classification | Gene involved and location | Inheritance | TSAT | Other clinical features |
|---|---|---|---|---|
| Type 1 | HFE; chr. 6 | AR | Increased | Adult-onset; more severe in males; highly variable clinical expression, with predominant liver damage and arthritis |
| Type 2A | HJV (hemojuvelin); chr. 1 | AR | Increased | Earlier onset (eg, <30 y old); similar severity in both sexes; prevalent cardiac and endocrine involvement |
| Type 2B | HAMP (hepcidin); chr. 19 | AR | Increased | Earlier onset (eg, <30 y old); similar severity in both sexes; prevalent cardiac and endocrine involvement |
| Type 3 | TFR2 (transferrin receptor 2); chr. 7 | AR | Increased | Very rare (look for parental consanguinity); clinically similar to type 1, with an earlier onset |
| Type 4A | SLC40A1 (ferroportin); chr. 2 | AD | Low-normal | Adult-onset; IO in the spleen; mild anemia; possible low tolerance to venesection |
| Type 4B | SLC40A1 (ferroportin); chr. 2 | AD | Increased | Very rare; in general, clinically similar to type 1, but more severe/early onset forms are reported |
| Classification | Gene involved and location | Inheritance | TSAT | Other clinical features |
|---|---|---|---|---|
| Type 1 | HFE; chr. 6 | AR | Increased | Adult-onset; more severe in males; highly variable clinical expression, with predominant liver damage and arthritis |
| Type 2A | HJV (hemojuvelin); chr. 1 | AR | Increased | Earlier onset (eg, <30 y old); similar severity in both sexes; prevalent cardiac and endocrine involvement |
| Type 2B | HAMP (hepcidin); chr. 19 | AR | Increased | Earlier onset (eg, <30 y old); similar severity in both sexes; prevalent cardiac and endocrine involvement |
| Type 3 | TFR2 (transferrin receptor 2); chr. 7 | AR | Increased | Very rare (look for parental consanguinity); clinically similar to type 1, with an earlier onset |
| Type 4A | SLC40A1 (ferroportin); chr. 2 | AD | Low-normal | Adult-onset; IO in the spleen; mild anemia; possible low tolerance to venesection |
| Type 4B | SLC40A1 (ferroportin); chr. 2 | AD | Increased | Very rare; in general, clinically similar to type 1, but more severe/early onset forms are reported |
AD, autosomal dominant; AR, autosomal recessive; chr., chromosome.
Poor applicability in clinical practice (limitations due to costs and lack of widespread expertise)
Apart from the genetic test looking for the common HFE variants, the identification of the molecular defect causing rarer forms of HC is currently offered by few laboratories, heterogeneously distributed and scattered worldwide. This requires that patients should travel, or DNA should be sent to referral centers, with inevitable discomforts, delays, and costs. Moreover, although the second-level genetic test (mainly based on NGS approach) has recently improved, with gradually decreasing costs, it demands advanced experience for a rigorous interpretation, which can take several weeks.14,87-89 Moreover, some cases of HC still remain molecularly undiagnosed even after NGS, suggesting the possibility of an unknown gene(s) yet to be discovered.89-91 For this reason, the cooperation between geneticists, bioinformaticians, and clinicians is necessary to resolve the most difficult cases. EuroBloodNet, the network connecting experts on rare hematological diseases, is making great efforts in this direction (for details, see www.eurobloodnet.eu).
Numerical subtypes do not capture the complex molecular pathogenesis of HC
Recent applications of NGS have highlighted that some patients with a provisional diagnosis of non-HFE HC cannot be ascribed to any of the numerical subtypes listed in the previous classification (Table 3). Essentially there are 2 reasons:
- 1.
Some show a “digenic” inheritance, deriving from the combination of pathogenic variants in 2 different genes involved in iron metabolism (eg, single p.Cys282Tyr + heterozygous variants in HJV, HAMP, or TFR2).90,92-95 Although there are still only few cases reported, digenic inheritance must also be considered in cases whose HFE genotype per se does not fully explain the clinical picture, for example, in patients with p.Cys282Tyr homozygosity and very early/severe IO.
- 2.
Others do not display variants in any of the 5 classical hemochromatosis genes (ie, HFE, HAMP, HJV, TFR2, and SLC40A1). Recently, some small case series96-98 have reported moderate late-onset IO in patients carrying variants in the BMP6 gene, encoding one of the major activators of hepcidin expression in response to iron.99 The role of such variants is still controversial, as they have been detected mostly in patients with a substantial burden of acquired cofactors.100 Nonetheless, they broaden the spectrum of genetic defects potentially responsible for HC.
Former type 4A HC
Former type 4A HC actually represents an IO syndrome characterized by distinctive clinical, biochemical, and pathological features which do not fit the definition of HC.101,102 They include normal to low TSAT, iron retention in spleen and hepatic macrophages, and, sometimes, poor tolerance to standard phlebotomies. The underlying molecular defect is the presence of loss-of-function (LOF) variants in the SLC40A1 gene that reduce expression or iron export capability of ferroportin at the cell surface. Therefore, iron is trapped inside iron-recycling macrophages (primarily in the spleen), resulting in a reduction of circulating iron and a tendency to iron-restricted erythropoiesis. The corresponding clinical features are normal-to-low TSAT and, sometimes, the development of mild anemia after phlebotomies. Both these elements are clearly at variance with the case definition of HC according to Table 1. Another peculiarity is represented by autosomal dominant inheritance. Taking into consideration all these aspects, many authors have suggested adopting a specific terminology for this condition, such as ferroportin disease (FD).14,102 In spite of very high ferritin levels which may be evident even in young-adult subjects, FD phenotype is generally milder than in HFE-related HC, possibly because of the lower toxicity of iron accumulation in macrophages as compared with hepatocytes.102,103
On the other hand, very rare gain-of-function (GOF) variants in the SLC40A1 gene lead to ferroportin resistance to hepcidin and cause IO conditions phenotypically and biochemically indistinguishable from hepcidin-deficient HC (former type 4B HC).104 Variants that interfere with hepcidin binding to ferroportin and also impair ferroportin stability or ability to export iron are also possible, potentially leading to a mixed or intermediate phenotype variably influenced by age or environmental factors.
Former type 2 molecular subtypes are not always juvenile forms and vice versa
As mentioned before, the term juvenile hemochromatosis classically designates an early-onset (within the second or third decades of life), fully-expressed HC phenotype showing similar penetrance in both genders and a tendency to present with cardiac and endocrine dysfunctions.27,105 This phenotype is generally due to variants in HJV and HAMP genes causing much more severe iron hyperabsorption than the HFE mutations. However, recent studies have highlighted some age overlap at diagnosis between the various molecular subtypes of HC.72 Therefore, the term juvenile HC can be ambiguous if invariably attributed to variants in the HAMP and HJV genes because in some of these patients, the disease is diagnosed in adulthood. Similarly, the term can be misleading in HC patients with defects in genes other than HJV or HAMP, but with early-onset severe phenotypes.
New classification of HC proposed by the working group
As a result of the 2-step process described previously, the panelists propose a new classification of HC (shown in Table 4), addressing both clinical issues (thereby addressing the needs of general clinicians and subspecialists) and molecular precision. The emphasis on clinical features obviates the current challenges represented by second-level genetic testing for detecting rare variants in the HFE and non-HFE genes, which could lead to delayed diagnosis and treatment. When the criteria listed in Table 1 are fulfilled, the diagnosis of HFE-related HC can be made in the presence of p.Cys282Tyr homozygosity. If an appropriately investigated patient has an unequivocal HC phenotype without cofactors but is not a p.Cys282Tyr homozygote (and this includes compound p.Cys282Tyr and p.His63Asp heterozygosity or p.His63Asp homozygosity), a provisional diagnosis of “molecularly undefined” HC can be made, and phlebotomies started. In this case, quantification of the total amount of iron removed by phlebotomies will serve as an additional marker of IO. The panelists agree that, whenever possible, an accurate molecular characterization remains important in these patients, especially for cascade screening of asymptomatic siblings or other first-degree relatives. To this end, patients should be referred (or DNA should be sent) to a specialized center. Indeed, second-level genetic tests have limitations that include costs, time delay, and poor availability in certain regions and require a high level of expertise for interpretation. Based on NGS results, some cases could be reclassified into HFE-related, digenic, or non-HFE HC (as shown in Figure 2).
New classification of HC proposed by the working group
| Novel classification | Molecular pattern | Note |
|---|---|---|
| HFE-related | p.Cys282Tyr homozygosity or compound heterozygosity of p.Cys282Tyr with other rare HFE pathogenic variants106-109 or HFE deletion110 | Low penetrance; consider presence of host-related or environmental cofactors for IO In subjects with other HFE genotypes (eg, p.Cys282Tyr/His63Asp compound heterozygosity or p.His63Asp homozygosity) consider second-line genetic testing for rarer variants |
| Non-HFE-related | Rare pathogenic variants in “non-HFE” genes: • HJV-related • HAMP-related • TFR2-related • SLC40A1 (GOF)-related | Potentially, mutations in any hepcidin-regulatory gene may be causative (the effects of novel mutations should be confirmed through functional and epidemiological studies) Molecular subtypes characterization only at specialized centers, but the diagnosis of non-HFE related HC is sufficient to start phlebotomies at nonspecialized centers* |
| Digenic† | Double heterozygosity and/or double homozygosity/heterozygosity for mutations in 2 different genes involved in iron metabolism (HFE and/or non-HFE) | More commonly, p.Cys282Tyr mutation in HFE gene might coexist with mutation in other genes; rarely, both mutations involve non-HFE genes |
| Molecularly undefined | Molecular characterization (still) not available after sequencing of known genes (provisional diagnosis) | Patients should be referred (or DNA should be sent) to specialized centers |
| Novel classification | Molecular pattern | Note |
|---|---|---|
| HFE-related | p.Cys282Tyr homozygosity or compound heterozygosity of p.Cys282Tyr with other rare HFE pathogenic variants106-109 or HFE deletion110 | Low penetrance; consider presence of host-related or environmental cofactors for IO In subjects with other HFE genotypes (eg, p.Cys282Tyr/His63Asp compound heterozygosity or p.His63Asp homozygosity) consider second-line genetic testing for rarer variants |
| Non-HFE-related | Rare pathogenic variants in “non-HFE” genes: • HJV-related • HAMP-related • TFR2-related • SLC40A1 (GOF)-related | Potentially, mutations in any hepcidin-regulatory gene may be causative (the effects of novel mutations should be confirmed through functional and epidemiological studies) Molecular subtypes characterization only at specialized centers, but the diagnosis of non-HFE related HC is sufficient to start phlebotomies at nonspecialized centers* |
| Digenic† | Double heterozygosity and/or double homozygosity/heterozygosity for mutations in 2 different genes involved in iron metabolism (HFE and/or non-HFE) | More commonly, p.Cys282Tyr mutation in HFE gene might coexist with mutation in other genes; rarely, both mutations involve non-HFE genes |
| Molecularly undefined | Molecular characterization (still) not available after sequencing of known genes (provisional diagnosis) | Patients should be referred (or DNA should be sent) to specialized centers |
Provided that IO is confirmed by MRI. If this is not accessible, close monitoring of Hb level is needed to avoid the occurrence of anemia.
Caution is needed to interpret as digenic inheritance results from NGS outputs reporting several variants in gene panels. Whenever possible, strict criteria for defining pathogenic variants should be adopted and corroborated by family segregation and/or functional studies.
Based on all the above considerations, we suggest adopting a new, more workable classification of HC (shown in Table 4) capable of capturing the growing genetic complexity of HC highlighted by NGS. In fact, the type-numerical classification does not allow assignation of any subtypes to patients with complex genotypes deriving from variants in 2 genes (“digenic” HC), nor to those who remain undefined after sequencing of known HC genes.
Finally, the panelists agreed to abandon the current terminology of type 4A and 4B HC, related to LOF and GOF variants in the ferroportin gene, respectively. While type B is it should be definitively renamed as “ferroportin disease” and included in inherited rare disorders of iron metabolism other than HC. It is important to recall that ferroportin mutations are characterized by an autosomal dominant inheritance pattern with important implications for genetic testing of family members.
In summary, the novel classification proposed here is based on a pathophysiological cornerstone (hepcidin deficiency) and a distinct clinical/biochemical phenotype. It recognizes the difficulties of a complete molecular characterization and has the potential of being easily shareable between practicing physicians and referral centers. Avoiding any ambiguity is essential for clear and effective communication that will facilitate proper diagnosis and treatment of HC.
Acknowledgments
The authors thank Paul C. Adams, Edouard Bardou-Jacquet, Patricia Bignell, Barbara Butzeck, Clara Camaschella, Robert Evans, Robert Fleming, Tomas Ganz, Olivier Loréal, Giacomo Marchi, Elizabeta Nemeth, Antonello Pietrangelo, Alberto Piperno, John D. Ryan, Mayka Sanchez, Paulo Santos, Dorine W. Swinkels, and Heinz Zoller for critically reviewing and editing the manuscript.
Authorship
Contribution: P.B., D.G., G.P., M.U.M., and I.C. led the panel and conceived the manuscript; and D.G. and F.B. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
A complete list of members of the Nomenclature Committee of the BIOIRON Society is provided in “Appendix.”
Correspondence: Domenico Girelli, Department of Medicine, Section of Internal Medicine, Policlinico Giambattista Rossi, 37134 Verona, Italy; e-mail: domenico.girelli@univr.it.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
Appendix
The full list of members of the Nomenclature Committee of the International Society for the Study of Iron in Biology and Medicine (BIOIRON Society) and affiliations is as follows: Domenico Girelli, Fabiana Busti, and Giacomo Marchi, University of Verona and Azienda Ospedaliera Universitaria Integrata, Verona, Italy; Pierre Brissot, Institut NuMeCan, Rennes, France; Ioav Cabantchik, Hebrew University, Jerusalem, Israel; Martina U. Muckenthaler, University of Heidelberg and German Center for Lung Research, Heidelberg, Germany, and German Centre for Cardiovascular Research, Partner Site Heidelberg, Mannheim, Germany; Graça Porto, Universidade do Porto and Porto University Hospital Center, Porto, Portugal; Paul C. Adams, University of Western Ontario, London, ON, Canada; Edouard Bardou-Jacquet, University of Rennes, Rennes, France; Patricia Bignell, The Churchill Hospital, Oxford, United Kingdom; Barbara Butzeck, European Federation of Associations of Patients with Haemochromatosis, Croissy-sur-Seine, France; Clara Camaschella, San Raffaele Scientific Institute, Milan, Italy; Robert Evans and Dianne Prince, Haemochromatosis International (Hemochromatosis International: global alliance linking associations of patients with hemochromatosis worldwide [http://haemochromatosisinternational.org/]), Bratton Fleming, United Kingdom; Robert Fleming, Saint Louis University, St Louis, MO; Tomas Ganz and Elizabeta Nemeth, University of California, Los Angeles, Los Angeles, CA; Olivier Loreal, Nutrition Metabolisms and Cancer (NuMeCan) Institute, Rennes, France; Antonello Pietrangelo, Azienda Ospedaliero-Universitaria di Modena, Policlinico, Modena, Italy; Alberto Piperno, S Gerardo Hospital, Monza, Italy; John D. Ryan, Beaumont Hospital/Royal College of Surgeons in Ireland, Dublin, Ireland; Mayka Sanchez, Universitat Internacional de Catalunya, Sant Cugat del Valles, Barcelona, Spain Paulo Santos, Universidade Federal de São Paulo, São Paulo, Brazil; Dorine W. Swinkels, Radboud University Medical Center, Nijmegen, The Netherlands, and Sanquin Research and Blood Bank, Amsterdam, The Netherlands; and Heinz Zoller, Christian Doppler Laboratory on Iron and Phosphate Biology, Innsbruck, Austria.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal