


Abstract
Chronic hemolytic anemia and intermittent acute pain episodes are the 2 hallmark characteristics of sickle cell disease (SCD). Anemia in SCD not only signals a reduction of red cell mass and oxygen delivery, but also ongoing red cell breakdown and release of cell-free hemoglobin, which together contribute to a number of pathophysiological responses and play a key role in the pathogenesis of cumulative multiorgan damage. However, although anemia is clearly associated with many detrimental outcomes, it may also have an advantage in SCD in lowering risks of potential viscosity-related complications. Until recently, clinical drug development for SCD has predominantly targeted a reduction in the frequency of vaso-occlusive crises as an endpoint, but increasingly, more attention is being directed toward addressing the contribution of chronic anemia to poor outcomes in SCD. This article aims to explore the complex pathophysiology and mechanisms of anemia in SCD, as well as the need to balance the benefits of raising hemoglobin levels with the potential risks of increasing blood viscosity, in the context of the current therapeutic landscape for anemia in SCD.
Introduction
Anemia is a major global public health problem; it affects an estimated one-third of the world population and accounts for 9% of the worldwide disability.1 The burden of anemia includes poor birth outcomes, delayed child development, neurocognitive impairment, decreased work productivity, and increased risk of hospitalization and mortality.2,3 Although the overall prevalence of anemia has decreased over recent decades,1 prevalence of congenital anemias such as sickle cell disease (SCD) continues to rise with reductions in childhood mortality from infectious disease, increased life expectancy, and continued population growth in low- and middle-income countries.
SCD affects >300 000 annual births globally.4 It is caused by an abnormal hemoglobin S (HbS) that results from a point mutation in the β-globin gene leading to substitution of glutamic acid by valine in position 6 of the β chain of hemoglobin (βGlu6Val). In well-resourced countries, with the dramatic reductions in childhood mortality, SCD has now evolved into a chronic disease in adults.5
Polymerization of HbS and red blood cell (RBC) sickling underlie the 2 key characteristics of SCD, chronic hemolytic anemia and episodic acute vaso-occlusive pain, the latter frequently referred to as vaso-occlusive crises (VOCs). As acute pain is the most prominent sickle complication, SCD guidelines, clinical management, and drug development have rightly focused on the treatment and prevention of VOCs, with VOC frequency often considered an acceptable surrogate endpoint in SCD.6 However, sickle-related pain remains difficult to objectively capture and quantify, and several interventional clinical trials using VOC frequency as the primary endpoint have been unsuccessful.7-9 Further, as SCD patients age, the ongoing hemolytic anemia becomes an increasingly important contributor to the cumulative organ damage, and more evidence-based surrogate endpoints for clinical benefit in SCD are needed.6,10
Although correcting anemia can only be beneficial for the vast majority of the general population, doing so in individuals with SCD may carry potential risks, and the treatment of anemia in SCD therefore requires a more nuanced approach. However, consensus is still lacking in the definition of what a safe and clinically meaningful hemoglobin concentration is, in patients with SCD.11
Here, we review the complex pathophysiology of anemia in SCD, the detrimental as well as compensatory effects of anemia, and their implications for the use of existing therapies for anemia in SCD.
Detrimental effects of anemia in SCD
Anemia impacts the health of SCD individuals from childhood and continues to have an insidious detrimental effect on organ function and overall quality of life. Cardiopulmonary disease, sickle nephropathy, and cerebrovascular disease are the leading causes of morbidity and mortality in the adult SCD population12-14 and are all exacerbated by the chronic anemic state (Table 1). Tangible effects of anemia include frailty, decreased mobility, increased risk of hospitalization, and mortality in the elderly,2,15 with whom the older adult SCD population shares many risk factors. In sum, chronic anemia contributes to and accelerates physiological aging in SCD patients.
Detrimental effects associated with low hemoglobin level in sickle cell disease
| Organ system | Detrimental effects |
|---|---|
| Cardiac | Cardiomegaly, left ventricular hypertrophy, dilated cardiomyopathy, diastolic dysfunction12,18 |
| Pulmonary | Elevated tricuspid regurgitant jet velocity and pulmonary artery systolic pressure, pulmonary hypertension12,23,32 |
| Renal | Albuminuria, sickle nephropathy28,32 |
| Neurological | Increased TCD velocity, stroke, silent cerebral infarction, neurocognitive impairment, proliferative retinopathy, cerebral vasculopathy and moyamoya syndrome12,20,98 |
| Musculoskeletal | Bone infarction, osteonecrosis, chronic skin ulcers, low bone mineral density20,22,72 |
| General | Fatigue, weakness, disability, health-related quality of life99,100 |
| Organ system | Detrimental effects |
|---|---|
| Cardiac | Cardiomegaly, left ventricular hypertrophy, dilated cardiomyopathy, diastolic dysfunction12,18 |
| Pulmonary | Elevated tricuspid regurgitant jet velocity and pulmonary artery systolic pressure, pulmonary hypertension12,23,32 |
| Renal | Albuminuria, sickle nephropathy28,32 |
| Neurological | Increased TCD velocity, stroke, silent cerebral infarction, neurocognitive impairment, proliferative retinopathy, cerebral vasculopathy and moyamoya syndrome12,20,98 |
| Musculoskeletal | Bone infarction, osteonecrosis, chronic skin ulcers, low bone mineral density20,22,72 |
| General | Fatigue, weakness, disability, health-related quality of life99,100 |
The pathology of anemia in SCD can be considered under 2 main components: intravascular hemolysis and low hemoglobin concentration or red cell mass. The role of intravascular hemolysis in perpetuating multiorgan damage in SCD has been well documented16,17 and will not be reviewed in detail here. Instead, we focus on the contribution of low hemoglobin level in the pathogenesis of chronic organ damage in SCD.
Cardiopulmonary effects of chronic anemia
Typically, when hemoglobin level drops below 10 g/dL, cardiac output increases as a physiological compensation to maintain oxygen transport.18 Increased cardiac output is achieved through lowering systemic vascular resistance via local vascular adaptations triggered by hypoxia, including vasodilation of arterioles in hypoxic tissues, neovascularization or recruitment and perfusion of existing vessels to increase the vascular cross-sectional area, and arteriovenous shunting; the latter increases vascular conductance but at the expense of tissue oxygen delivery.12,18,19 These potentially maladaptive vascular responses to chronic hypoxia manifest classically in SCD as proliferative retinopathy, moyamoya syndrome, and other cerebral vasculopathy12,20 (Table 1).
Simultaneously, lower systemic vascular resistance and lower blood pressure cause neurohormonal activation, including activation of the sympathetic nervous system and the renin-angiotensin-aldosterone system, leading to increased vasoconstriction and decreased perfusion of tissue beds, nitric oxide synthase (NOS) inhibition, endothelial dysfunction, and vascular inflammation.18,19,21 This abnormal vascular regulation, in combination with mechanical obstruction of the microvasculature triggered by sickled RBCs, directly contributes to the development of complications such as sickle nephropathy and chronic skin ulcers.20,22
Over time, anemia-induced high cardiac output and chronic left ventricular (LV) volume overload lead to abnormalities in cardiac structure and pulmonary hemodynamics.14,23,24 This predominantly rheologic cardiomyopathy has been observed in individuals with other causes of chronic anemias, both congenital (eg, pyruvate kinase deficiency) and acquired (helminth infection and iron deficiency), and have been shown to be reversible with correction of the anemia.12 Similarly, in individuals with SCD, improvements in LV size and LV end-diastolic volume have been observed as early as 3 months following successful hematopoietic stem cell transplant.25
The compounding role of chronic intravascular hemolysis and vasculopathy
Compounding the physiological consequences of chronic anemia, intravascular hemolysis contributes to the development of sickle vasculopathy, which together with poor tissue oxygen delivery leads to ischemia and chronic organ damage. A number of pathways involving release of cell-free hemoglobin, free heme, arginase, and other products of hemolysis into the circulation have been proposed to drive vascular dysfunction in SCD and have been recently reviewed.16,17 Briefly, increased cell-free hemoglobin from intravascular hemolysis promotes scavenging of nitric oxide, inhibition of endothelial NOS signaling, and formation of reactive oxygen species, leading to endothelial dysfunction, impaired vasodilatory responses, sterile inflammation, and oxidative stress.17,26 High rates of intravascular hemolysis have been associated with a number of vasculopathy-related complications, including pulmonary hypertension, stroke, leg ulcers, priapism, and renal dysfunction, as well as increased mortality.17,20,27,28
Chronic vasculopathy in SCD impedes physiological responses to anemia. Impaired oxygen delivery from anemia is typically compensated through both increased cardiac output and increased oxygen extraction in tissues. In SCD, vasodilatory mechanisms for increasing blood flow and augmenting cardiac output are chronically activated at baseline, diminishing physiological reserve. Compared with healthy controls as well as individuals with nonsickle etiologies of chronic anemia,29 there appears to be impaired oxygen extraction in adults with SCD, which may potentially be related to the microvascular occlusion and damage to capillary beds, capillary wall thickening, arteriovenous shunting, or higher cerebral blood flow observed in SCD.12,29 Although HbS polymerization may paradoxically improve the efficiency of local oxygen delivery in hypoxic tissues as a result of the decreased Hb oxygen affinity, the effect is defeated by a subsequent increase in hemolysis and micro-vasoocclusion, which worsen the hypoxic state. The combination of impaired oxygen delivery, limited vasodilatory reserve, impaired oxygen extraction, and recurrent microvascular occlusions ultimately results in tissue ischemia and infarction, as is observed with silent cerebral infarcts.30,31
A recent meta-analysis found lower hemoglobin levels to be associated with stroke, silent cerebral infarcts, increased transcranial Doppler velocity, albuminuria, and elevated pulmonary artery systolic pressure in SCD and that a modeled hemoglobin increase of at least 1 g/dL predicted a 64% reduction in the risk of mortality.32 Other recent data suggest that treating anemia as an independent cause of morbidity can improve outcomes in SCD.33 However, concerns surrounding increasing blood viscosity by raising hemoglobin levels complicate the development of therapeutic approaches.11
Compensatory effects of anemia and the role of blood viscosity
The notion that higher hemoglobin levels in SCD may be detrimental as a result of increased blood viscosity arose from both laboratory and observational studies of RBC rheology and disease epidemiology, as well as anecdotal case reports.11,34-37 Numerous in vitro studies using sickle RBCs and microfluidics have attempted to simulate the in vivo situation in SCD, but defining the factors contributing to raising blood viscosity in SCD remains a challenge. Factors proposed include higher intracellular HbS percentage, deoxygenation, and lower shear rates,37,38 likely as a result of the reduced RBC deformability, increased density, and increased adhesiveness, which promote vaso-occlusion (Figure 1). Tissue-specific vascular autoregulation and vascular geometry may further modulate the effect of blood viscosity in vivo.12,39
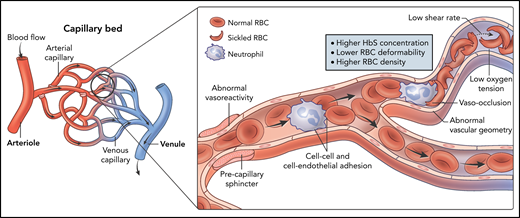
Determinants of blood viscosity in SCD. In vitro models have demonstrated higher whole blood viscosity in SCD blood samples compared with non-SCD samples. The viscosity of sickle blood increases with deoxygenation, low shear rates, and higher HbS concentration, which cause increased HbS polymerization and sickling. Less deformable and denser red cells, as well as increased cellular adhesivity, also raise the viscosity of sickle blood. Additionally, in vivo, vasculopathy and endothelial dysfunction may contribute to increased blood viscosity and vaso-occlusion in the microcirculation.
Determinants of blood viscosity in SCD. In vitro models have demonstrated higher whole blood viscosity in SCD blood samples compared with non-SCD samples. The viscosity of sickle blood increases with deoxygenation, low shear rates, and higher HbS concentration, which cause increased HbS polymerization and sickling. Less deformable and denser red cells, as well as increased cellular adhesivity, also raise the viscosity of sickle blood. Additionally, in vivo, vasculopathy and endothelial dysfunction may contribute to increased blood viscosity and vaso-occlusion in the microcirculation.
In vitro studies of RBC rheology in SCD have suggested a positive correlation between VOC frequency and either blood viscosity40,41 or RBC deformability,34,42,43 but the sample size of these studies is small, and negative studies exist as well.41,44 A few clinical studies have associated higher hemoglobin or hematocrits with increased frequency of painful crises,34-36 but comorbidities were not accounted for, and these studies were limited by their retrospective nature. Nonetheless, these studies led to the notion that anemia may be protective and play a compensatory role by reducing blood viscosity and microvascular occlusion in SCD. This hypothesis, in conjunction with the risks of alloimmunization and hemolytic transfusion reactions associated with blood transfusions, may explain why chronic anemia in SCD is not treated as aggressively as in other hematological disorders, such as thalassemia and myelodysplastic syndromes, where higher hemoglobin targets have been shown to reduce morbidity and mortality and improve quality of life.45,46
However, there is limited data to inform whether and how these observations of abnormal blood rheology could impact clinical management in SCD. Clinical observations have noted that SCD individuals with lower hemoglobin levels and a higher hemolytic rate appear to have fewer painful crises and a disease phenotype dominated by vasculopathic complications, such as leg ulcers, stroke, priapism, and pulmonary hypertension.17,27,47 In contrast, it has been suggested that as a result of increased blood viscosity, individuals with higher hemoglobin levels have an increased risk of complications associated with microvascular occlusion, such as acute pain, acute chest syndrome, and osteonecrosis, a phenotype also associated with coinheritance of α-thalassemia.17,47
Evaluating hemoglobin targets
The development of new therapies impacting anemia in SCD provide an opportunity to re-examine hemoglobin targets in SCD that must take into account concerns about raising blood viscosity. A phase 3 clinical trial of the Gardos channel inhibitor senicapoc found that the senicapoc-only arm achieved higher hemoglobin levels but also had a higher rate of VOC,7 raising safety concerns about higher hemoglobin levels leading to more viscosity-related vaso-occlusion. However, a recent re-analysis of subjects with hemoglobin response vs placebo showed no significant difference in VOC rate,48 suggesting that changes in absolute hemoglobin level may not be a useful biomarker for predicting adverse outcomes related to blood viscosity in vivo. Indeed, vascular function and compensation may play a large part in determining whether an increase in hemoglobin in an individual is beneficial or detrimental. Several studies have suggested that higher vessel wall shear stress from increased blood viscosity can stimulate production of nitric oxide from the vascular endothelium, resulting in vasodilation and improved microvascular blood flow and tissue perfusion.39,49,50 However, individuals with endothelial dysfunction associated with SCD may have impairment in normal vascular adaptations, leading to reduced blood flow with increasing blood viscosity.51
Another important factor to consider is the speed of correction of the anemia, especially in the setting of inadequate vascular compensation. Anecdotally, rapid correction of anemia with blood transfusions has led to devastating neurological sequelae in individuals with SCD.11,52 The sudden increase in blood viscosity from transfusions is thought to have a detrimental effect similar to that of increased serum immunoglobulins or leukostasis in causing hyperviscosity syndrome, with sludging of the blood, decreased microvascular blood flow, and resultant tissue ischemia. To prevent viscosity-associated complications in SCD, transfusion guidelines recommend avoiding raising hemoglobin levels above 10 g/dL. However, there is limited clinical data to support this absolute threshold and no clear guidance on an appropriate rate of hemoglobin correction.11
It is important to note that the composition of hemoglobin is critical when considering hemoglobin thresholds. Transfusion, whether simple of exchange, retains a population of sickle RBCs, which contain predominantly HbS. In simple transfusion therapy, reducing the percentage of sickle RBCs is limited by the final absolute hemoglobin level to avoid triggering vaso-occlusive complications. Exchange transfusion replaces sickle RBCs with nonsickle donor RBCs with the aim of lowering the sickle RBC population and is less constrained by the final absolute hemoglobin level. Chronic regular exchange transfusion therapy can achieve slow gradual hemoglobin increases, concurrently lowering HbS percentage and allowing patients to be safely maintained at hemoglobin concentrations of 12 to 13 g/dL without causing viscosity-related issues.11 Similarly, hematopoietic stem cell transplantation and genetic therapies dramatically lower endogenous production of HbS, shifting hemoglobin composition toward hemoglobin A and reducing the contribution of HbS polymerization to blood viscosity. As such, SCD patients have tolerated partial or full correction of their anemia postcellular therapy without increased vaso-occlusive or viscosity-related complications.11 Therefore, it is not likely to be feasible to set a universally “safe” hemoglobin threshold in SCD as optimal hemoglobin targets are dependent upon multiple factors, including the mechanism of anemia correction and hemoglobin composition, the rate of hemoglobin correction, and individual differences in organ and vascular function.
Monitoring treatments for anemia
Balancing the potential risks of increasing blood viscosity with the known detrimental effects of chronic anemia is critical (Figure 2) and requires careful monitoring. Cohort studies and clinical trials of therapies affecting hemoglobin level should strive to include rheological measurement such as whole blood viscosity and RBC deformability as biological endpoints as these parameters will provide investigators with a more complete picture of both drug effects and potential SCD subphenotypes. Although there have been recent efforts to incorporate ektacytometry into novel drug studies,53,54 clinical trials have yet to report in vivo drug effects on blood viscosity. The adoption of a comprehensive laboratory-based rheological profile, coupled with ex vivo and in vivo techniques of quantifying microvascular blood flow, such as microfluidics and near-infrared spectroscopy, would provide novel and powerful approaches for understanding how anemia correction may be done safely in patients with SCD.
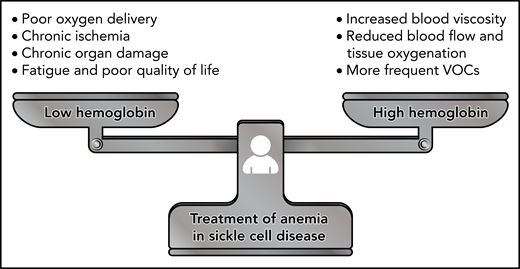
Finding the balance in the treatment of anemia in SCD. The correction of anemia in SCD requires careful balancing of the detrimental effects of anemia with the potential risks associated with increased blood viscosity. Anemia is known to impair oxygen delivery, contribute to chronic ischemia and organ damage, and affect patients’ quality of life. On the other hand, there is limited evidence for whether raising hemoglobin levels through transfusion or drug therapy can lead to increased vaso-occlusive pain, reduced tissue perfusion, and viscosity-associated complications such as stroke. Additional work is needed to identify safe hemoglobin targets and balanced treatment approaches for managing anemia in SCD.
Finding the balance in the treatment of anemia in SCD. The correction of anemia in SCD requires careful balancing of the detrimental effects of anemia with the potential risks associated with increased blood viscosity. Anemia is known to impair oxygen delivery, contribute to chronic ischemia and organ damage, and affect patients’ quality of life. On the other hand, there is limited evidence for whether raising hemoglobin levels through transfusion or drug therapy can lead to increased vaso-occlusive pain, reduced tissue perfusion, and viscosity-associated complications such as stroke. Additional work is needed to identify safe hemoglobin targets and balanced treatment approaches for managing anemia in SCD.
Factors contributing to anemia in SCD
Increased RBC breakdown
Extravascular and intravascular hemolysis are well recognized mechanisms of anemia in SCD, reducing sickle RBC lifespan from an average of 120 to 7-14 days.55 Extravascular hemolysis may be the dominant form of RBC destruction, with abnormal phosphatidylserine exposure and reduced deformability of sickled RBCs prompting clearance by macrophages in the reticulo-endothelial system (Figure 3). Several mechanisms likely contribute to intravascular hemolysis in SCD, including increased RBC rigidity and membrane fragility, complement-mediated lysis facilitated by phosphatidylserine exposure, and oxidative membrane damage.17 There is a lack of consensus about relative contributions of extravascular vs intravascular hemolysis, and the balance may be variable between individuals. A number of approved and investigational drugs target the reduction of intravascular hemolysis, predominantly through antisickling mechanisms, and have been reviewed elsewhere.56,57 Splenectomy, which may be performed to reduce extravascular hemolysis in other hemolytic disorders (eg, hereditary spherocytosis), is rarely used as a treatment modality in SCD outside of splenic complications, such as recurrent splenic sequestration.
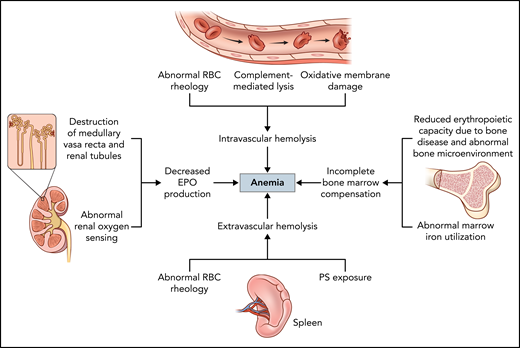
Factors contributing to anemia in SCD. The main mechanisms driving anemia in SCD include intravascular and extravascular hemolysis, decreased EPO production, and incomplete bone marrow compensation. Different mechanisms may predominate or act synergistically in different individuals with SCD, contributing to the phenotypic variability of the disease. Using a combination of targeted therapies to address more than 1 mechanism simultaneously may provide more effective and individualized approaches to treating anemia in SCD.
Factors contributing to anemia in SCD. The main mechanisms driving anemia in SCD include intravascular and extravascular hemolysis, decreased EPO production, and incomplete bone marrow compensation. Different mechanisms may predominate or act synergistically in different individuals with SCD, contributing to the phenotypic variability of the disease. Using a combination of targeted therapies to address more than 1 mechanism simultaneously may provide more effective and individualized approaches to treating anemia in SCD.
Impaired erythropoietic response due to inappropriately low EPO production
In healthy individuals, the reduction of RBC mass due to RBC destruction and resultant hypoxia lead to increased erythropoietin (EPO) production by the oxygen-sensing peritubular interstitial fibroblast in the renal cortex. EPO, a multifunctional cytokine, then drives erythropoiesis by promoting early erythroid progenitor commitment and entry into the cell cycle, as well as preventing apoptosis and promoting terminal differentiation of late progenitors.58 However, there is incomplete erythropoietic compensation for the chronic anemia in SCD due to a combination of factors (Figure 3), including renal dysfunction. A retrospective study showed that SCD patients have lower EPO levels for similar degrees of anemia compared with non-SCD individuals, and a sharp rise in EPO levels was not observed in SCD patients until hemoglobin levels dropped below around 9 g/dL, compared with around 12 g/dL in individuals with nonhemoglobinopathy-related anemia. An even more blunted EPO response was observed in adults compared with pediatric patients with SCD,59 suggesting that EPO production may decline as progressive renal dysfunction accumulates with age. Indeed, the development of sickle nephropathy begins in early childhood, manifesting as urinary concentrating defects, enuresis, glomerular hyperfiltration, progressive proteinuria, and eventually chronic kidney disease (CKD).60
Impaired erythropoietic response due to abnormalities of the bone and bone marrow niche
Iron depletion and abnormal iron utilization likely also contribute to inadequate erythropoietic drive, though bone marrow function in SCD is still poorly understood. Previous studies of iron stores in anemic adults and children with SCD have shown that one-quarter to one-half of SCD individuals have absent or diminished marrow iron staining.61,62 Interestingly, one study found that mean serum ferritin levels were >500 ng/mL in SCD subjects both with and without stainable marrow iron, suggesting that the lack of marrow-stainable iron may be caused by poor iron mobilization rather than true iron deficiency.63 In this sense, the impaired erythropoietic response in SCD may parallel the anemia of chronic inflammation in CKD, with impaired iron utilization resulting from the systemic chronic inflammatory state evidenced in SCD patients.16,26
Although a lack of marrow iron would be expected to reduce erythropoietic capacity and worsen anemia, other data suggest that iron deficiency may actually be protective in SCD in lowering intracellular HbS concentration.61,64-67 Iron restriction in the SCD mouse model has led to improvement in anemia, reduction of hemolysis, decreased mean corpuscular hemoglobin concentration and severity of sickling, and improved RBC deformability.68,69 In case reports, overt iron deficiency in SCD patients has also been associated with a decrease in the frequency of pain crises, but stronger evidence of clinical benefit is lacking.64,65
The insufficient EPO production and iron-restricted erythropoiesis in SCD are compounded by intrinsic bone marrow hyporesponsiveness, likely resultant from chronic ischemic damage from vaso-occlusion and abnormal bone marrow vascular architecture from pathologic angiogenesis.70 Anemia itself worsens bone health by inducing bone marrow hyperplasia and hypoxia-inducible factor–mediated EPO production, which stimulates osteoclast precursors and leads to bone loss.71,72 Disruption of the supportive bone microenvironment may in turn impair erythropoiesis,71 further inhibiting normal erythropoietic responses to anemia (Figure 3). In an animal model of SCD, the abnormal marrow neovascularization resulting from vaso-occlusion was completely reversed with blood transfusion targets of <30% HbS,70 raising the question of whether transfusion and other antisickling therapies can improve long-term bone marrow health and erythropoietic response in SCD.
Existing therapies for anemia in SCD
Treatment options for anemia in SCD to date have been limited and suboptimal (Table 2). Blood transfusions are the mainstay of treatment of severe or acute episodes of anemia, severe vaso-occlusive events, stroke prevention, and prevention of perioperative complications, but it is not typically used to correct chronic anemia in patients with stable SCD unless otherwise indicated for comorbidities such as pulmonary hypertension.73 There is growing hesitancy surrounding the liberal use of transfusion therapy due to increasing recognition of the risks of hemolytic transfusion reactions, alloimmunization, and iron overload.74 Transfusion as a strategy for addressing chronic anemia is also limited in low-resource settings by the extensive blood banking support needed to ensure safe and compatible blood products.
Comparison of existing therapies for anemia in SCD
| Blood transfusions | Hydroxyurea | Erythropoiesis-stimulating agents | Voxelotor | |
|---|---|---|---|---|
| Indications |
|
|
|
|
| Limitations |
|
|
|
|
| Blood transfusions | Hydroxyurea | Erythropoiesis-stimulating agents | Voxelotor | |
|---|---|---|---|---|
| Indications |
|
|
|
|
| Limitations |
|
|
|
|
Hydroxyurea was the first drug approved for and remains the only drug with demonstrated mortality benefit in SCD. Patients treated with hydroxyurea may reach hemoglobin levels above 10 g/dL without experiencing viscosity-related complications, possibly because of the antisickling effect of hemoglobin F (HbF) induction and improvements in RBC rheology.11 However, hydroxyurea’s effect on ameliorating anemia is modest, particularly in the adult population,75,76 and chronic anemia is not an indication for initiating hydroxyurea therapy, though some have proposed its use as a blood-conserving strategy during COVID-19 pandemic.77,78
Erythropoiesis-stimulating agents (ESAs), or exogenous EPO, are the standard of care for treating anemia in CKD. The inappropriately low EPO production and high prevalence of renal dysfunction in SCD provide a strong rationale for the use of ESAs. In addition, ESAs may have nonhematological cardioprotective and neuroprotective effects during ischemic injury, possibly related to activation of endothelial NOS or NF-κB signaling, as well as potential anti-inflammatory properties,79 which could potentially reduce organ damage from repeated cycles of vaso-occlusion. However, data on the efficacy and safety of ESAs in SCD are limited to small studies showing conflicting results.80-85 Older small case series investigated the potential use of ESAs to induce a HbF response alone or in combination of hydroxyurea but found inconsistent responses,81,84,85 whereas a more recent retrospective study did observe increases in HbF and hemoglobin level with treatment.83 Two other retrospective studies found an increase in hemoglobin level and/or decrease in transfusion burden after ESA therapy, without increased rates of VOC or thrombosis,80,82 suggesting that ESAs may be safe and efficacious in patients with SCD. On the other hand, there are anecdotal and rare case reports of ESAs provoking pain crises in SCD patients,81,83 especially in those not on hydroxyurea. Additionally, as patients with SCD may require much higher doses of ESAs to achieve a hemoglobin response, the cardiovascular and thrombotic risks associated with use of high-dose ESAs in CKD must be considered.86-88 Without additional evidence of efficacy and safety and standardization of dosing, ESAs will likely continue to be prescribed ad hoc by providers and underutilized as a treatment option for chronic anemia in SCD.
New and emerging therapies for anemia in SCD
In recent years, the drug therapeutic landscape for SCD has experienced exponential growth, culminating in the Food and Drug Administration approval of l-glutamine (Endari) in 2017 as a second drug therapy for SCD. This was quickly followed by the approval of 2 additional SCD-specific therapies, voxelotor (Oxbryta) and crizanlizumab (Adakveo) in 2019, and the most recent approval of deferiprone (Ferriprox) for transfusional iron overload in SCD and other anemias. The breadth of drug and cellular therapeutic development has been covered thoroughly in recent reviews56,57,89 and is not the focus of this article.
Although the vast majority of clinical trials have aimed to reduce vaso-occlusive complications, few have focused on the expected improvement in anemia with reduction of sickling and hemolysis. The recent accelerated Food and Drug Administration approval of voxelotor points to the unmet need for therapeutic approaches for anemia in SCD. Voxelotor is the only drug therapy approved on the basis of raising hemoglobin levels in SCD, with a hemoglobin increase of >1 g/dL in over half of patients treated at the higher dose level.90 Voxelotor works by shifting the p50 of hemoglobin, decreasing the oxygen tension at which sickle RBCs sickle, and inhibiting HbS polymerization. In vitro data also suggest that voxelotor can increase RBC deformability and reduce blood viscosity,54 which may help balance out the increase in red cell mass and avoid viscosity-related complications. However, its mechanism of action of increasing hemoglobin oxygen affinity has raised concerns about its effects on tissue oxygen delivery.11,91-93 Furthermore, unlike with hydroxyurea, trials of voxelotor have not yet demonstrated an impact on clinical outcomes such as VOC frequency and health-related quality of life.94
New classes of drugs are under development for SCD must grapple with these questions of how best to treat anemia while balancing safety and efficacy. The drug class farthest along in development is the red-cell pyruvate kinase (PKR) activators etavopivat (FT-4202) and mitapivat (AG-348), currently in phase 1 to 3 trials as a treatment of pyruvate kinase deficiency, thalassemia, and SCD. By increasing flux through the glycolytic pathway, PKR activators decrease red cell 2,3-DPG and increase adenosine triphosphate levels.95 In addition to the antisickling effect of lowering 2,3-DPG, increasing adenosine triphosphate levels may also prevent RBC dehydration through potassium loss via the Gardos channel, improving RBC membrane integrity and deformability. Indeed, PKR activators appear to improve the rheology of sickle RBCs ex vivo and in mouse models.96,97 Though PKR activators would be expected to decrease hemolysis and increase hemoglobin level through shifting hemoglobin oxygen affinity similar to voxelotor, the potential improvements in RBC health may theoretically counterbalance the left-shift in oxygen affinity, reminiscent of the pleotropic effects of hydroxyurea. Nevertheless, it remains to be seen what long-term benefits these and other novel therapies can offer SCD patients in comparison with the standard hydroxyurea therapy. In addition, questions of how we monitor and define “safe” hemoglobin thresholds that confer maximum clinical benefits with minimal risks, and how this may differ between patients, remain unanswered and require further investigation.
Conclusion
Chronic anemia is an increasingly recognized independent contributor to morbidity, premature mortality, and poor quality of life in SCD. However, anemia in SCD is complex and multifaceted. Treating anemia as an entity in itself in SCD is a delicate balancing act in which raising levels of hemoglobin may address detrimental effects of chronic anemia, but normalization of hemoglobin level may increase blood viscosity and paradoxically worsen SCD pathophysiology. As more experience is gained with existing and emerging novel drug therapies, we may see a shift toward the use of multimodal drug therapy to simultaneously reduce vaso-occlusion, raise hemoglobin levels, and improve RBC rheology, offering a safer and more effective approach to addressing chronic anemia in SCD.
Acknowledgments
This work was supported by the Division of Intramural Research of the National Heart, Lung, and Blood Institute, National Institutes of Health. Figure illustrations by Ethan Tyler and Alan Hoofring, National Institutes of Health.
Authorship
Contribution: J.Z.X. developed the ideas and wrote the first draft of the manuscript; and S.L.T. wrote and edited the manuscript.
Conflict-of-interest disclosure: S.L.T. is leading investigator-initiated trials of mitapivat (AG-348) in patients with sickle cell disease. J.Z.X. declares no competing financial interests.
Correspondence: Julia Zhe Xu, Section of Benign Hematology, Division of Hematology/Oncology, Department of Medicine, University of Pittsburgh Medical Center, 200 Lothrop St, Pittsburgh, PA 15213; e-mail: xujz2@upmc.edu.

Comments
Anemia, Pregnancy, and Sickle Cell Disease: Balancing on a Tightrope
Our perspective article is meant to provide an overview of the main etiologies, the primary physiological changes, and the overall deleterious effect of anemia in SCD, without discussing specific complications or management situations. The challenges of anemia management in pregnancy may be more appropriately addressed in a discussion of blood transfusion in SCD, or perhaps in a review on pregnancy in SCD. We agree that anemia in pregnancy deserves special mention, considering the significant morbidity associated with anemia and maternal and fetal outcomes in SCD, as well as the lack of disease-modifying therapies aside from transfusions with known or acceptable safety profiles in pregnancy.
Additionally, the question of how anemia in pregnancy influences vascular function, hemorheology, and cardiovascular reserve in individuals with SCD is compelling and deserves further investigation. The compounding of detrimental effects of anemia on top of pregnancy and SCD physiology may indeed underlie some of the poor maternal and fetal outcomes observed. At the same time, both pregnancy and SCD are associated with hypercoagulability and vascular dysfunction; pregnancy in SCD is also associated with increased risk of venous thromboembolism and pre-eclampsia.2-5 As such, extra caution and consideration of the opposing pathophysiological processes at play should be considered when addressing anemia in this population, especially with the use of simple blood transfusion, where dramatic increases in blood viscosity could potentially exacerbate vaso-occlusion, thrombophilia, hypertension, or even risk of stroke associated with eclampsia. Given the paucity of evidence, prospective studies, registries, and use of real-world data can greatly improve our understanding of the burden of anemia, as well as the potential therapeutic role of anemia correction in preventing pregnancy-related complications in SCD. Indeed, a clinical study evaluating maternal and fetal outcomes in pregnant women with SCD treated with serial prophylactic exchange blood transfusion compared to standard care is ongoing (ClinicalTrials.gov: NCT03975894), and additional studies evaluating the management of anemia and pregnancy in SCD are desperately needed.
Anemia in Sickle Cell Disease: The placenta is an end organ
Antenatal anemia is associated with maternal and fetal complications including stillbirth and intra-uterine growth restriction in the general population.1 Mechanistic and clinical evidence suggest anemia may also negatively affect SCD pregnancies. The angiogenic signaling molecules that are associated with abnormal placenta formation and maturation are over-expressed in chronic anemia in SCD.2 Clinically, a low first trimester hemoglobin is identified as a predictor for adverse maternal events in SCD pregnancy.3 We do not yet know whether people with chronic anemia from SCD, who have endothelial dysfunction, increased cardiac output, and decreased systemic vascular resistance at baseline, have the cardiovascular reserve to adapt to the additional strain of pregnancy.
Evidence suggests a modest benefit of prophylactic transfusions in pregnancy.3 However, indications for initiation of this therapy are indiscriminate and do not consider anemia risks.4 This may be partly explained by the near-universal presence of anemia in SCD pregnancy and the lack of a transfusion threshold below which maternal or fetal morbidity would be expected.
Besides transfusion, no SCD treatments are established in pregnancy. Hydroxyurea is a chemotherapeutic agent and its discontinuation at conception is recommended, although the lack of alternatives to transfusions leads to occasional use during pregnancy. Neither voxelotor nor crizanlizumab had promising pregnancy safety profiles in animal studies, and there is no data on L-glutamine.5
Reproductive organs are the less often considered end-organs in SCD. However, patients around the globe are surviving into their reproductive years. Continued research on the effects of anemia in SCD on all reproductive end-organs, including the placenta and fetus, is imperative.