

In this issue of Blood, 1 construct a comprehensive atlas of the endothelial-to-hematopoietic transition (EHT) continuum, as well as the subaortic niche cells in mouse embryonic aorta using a set of hemogenic endothelium (HE) reporter models. The atlas was then used to identify a novel surface marker of the HE continuum and decipher the precise cellular and molecular changes that are caused by deficiency of the pivotal transcription factors Runx1b and Gfi1/Gfi1b (see figure).
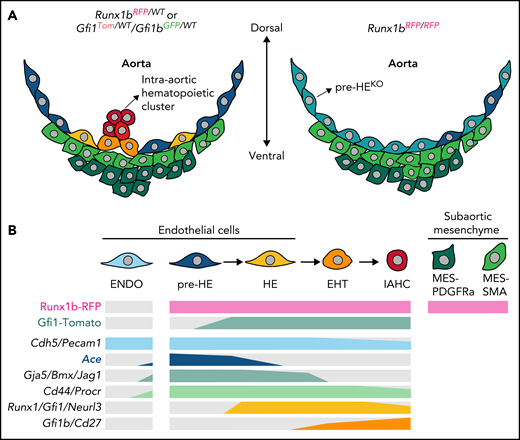
Molecular changes caused by deficiency of Runx1b and Gfi1/Gfi1b. (A) Cellular constitution of the ventral part of the aortic structure where HSCs are generated in midgestational mouse embryos: Runx1b or Gfi1/Gfi1b heterozygous embryo (left panel) and Runx1b homozygous-knockout (KO) embryo (right panel). Runx1b deletion leads to an extensive change in pre-HE characteristics in addition to the loss of HE, EHT, and intra-aortic hematopoietic cluster cells, as well as alteration of the subaortic niche composition. (B) Expression of the reporters (top 2 lines) and the transcripts of representative markers (lower 6 lines) in different cell populations, including EHT continuum. Gray color indicates very low expression. Note the specific expression of Ace in pre-HE. ENDO, endothelial; IAHC, intra-aortic hematopoietic clusters; MES-PDGFRa, PDGFRa+ mesenchymal; MES-SMA, SMA+ mesenchymal; WT, wild-type. The figure was created by Siyuan Hou (Jinan University, Guangzhou, China).
Molecular changes caused by deficiency of Runx1b and Gfi1/Gfi1b. (A) Cellular constitution of the ventral part of the aortic structure where HSCs are generated in midgestational mouse embryos: Runx1b or Gfi1/Gfi1b heterozygous embryo (left panel) and Runx1b homozygous-knockout (KO) embryo (right panel). Runx1b deletion leads to an extensive change in pre-HE characteristics in addition to the loss of HE, EHT, and intra-aortic hematopoietic cluster cells, as well as alteration of the subaortic niche composition. (B) Expression of the reporters (top 2 lines) and the transcripts of representative markers (lower 6 lines) in different cell populations, including EHT continuum. Gray color indicates very low expression. Note the specific expression of Ace in pre-HE. ENDO, endothelial; IAHC, intra-aortic hematopoietic clusters; MES-PDGFRa, PDGFRa+ mesenchymal; MES-SMA, SMA+ mesenchymal; WT, wild-type. The figure was created by Siyuan Hou (Jinan University, Guangzhou, China).
Hematopoietic stem cells (HSCs) are widely used in clinical treatment. Considering their limited numbers, generating HSCs in vitro has always been a major goal of regenerative medicine. The importance of a clear understanding of the cellular evolution and molecular program of HSC generation during development cannot be overstated. HSCs originate from vascular endothelial cells located predominantly in the ventral side of the aorta in midgestational mouse embryos. The transient and rare vascular endothelial cells with the ability to produce hematopoietic cells are called HE. The HSC-primed HE will form pre-HSCs localized mainly within the intra-aortic clusters through the EHT.2
To be able to isolate HE for further study, researchers have discovered several markers and established corresponding reporters to enrich HE, represented by Runx1, Gfi1, and newly identified Neurl3.3-5 However, a single surface marker for HE enrichment in lieu of transgenic animal models has not been identified. The roles of Runx1 and its downstream target Gfi1 are well recognized; Runx1 is required for EHT, but not thereafter, and Gfi1 participates in the loss of endothelial properties during this process.3,6 However, the precise cellular changes and molecular events that occur in the absence of these important transcription factors have not been deciphered at the whole transcriptomic level. The above issues are addressed in the study by Fadlullah et al.
In recent years, a series of studies at the single-cell level, involving single-cell transcriptomics and single-cell functional assays, have revealed the initial fate choice of HE from upstream arterial endothelium, uncovered the stepwise developmental path from HE to HSCs, identified several important intermediate cell populations, and revealed the molecular events underlying this EHT trajectory.4,5,7,8 Fadlullah et al took this a step farther and isolated a series of phenotypic HE-related populations from the midgestational aorta region using heterozygous and homozygous Runx1b and Gfi1/Gfi1b reporter mouse models to perform high-precision full-length single-cell RNA sequencing (scRNA-seq) of nearly 1200 cells. The data set shows greater gene detection sensitivity than do previous studies using similar phenotypic populations.5,8
The investigators identified several clusters encompassing the whole EHT continuum, which were respectively annotated as pre-HE, HE, EHT, and intra-aortic hematopoietic cluster cells. Importantly, by profiling the cells from homozygous mice with both alleles replaced by the reporter, the exact molecular changes during the EHT procedure resulting from Runx1b or Gfi1/Gfi1b deficiency were revealed. Runx1b deletion led to obstruction of pre-HE to HE differentiation, consistent with a previous finding showing that Runx1 dosage regulates the efficiency of pre-HE to HE transition.5 Specifically, the homozygous deletion of Runx1b resulted in the appearance of a distinct pre-HE population with unique characteristics. On the other hand, the deletion of Gfi1/Gfi1b led to the inability of HE to develop into EHT cells, with the Gfi1/Gfi1b-deficient HE diverted away from the canonical EHT developmental trajectory. In addition, by analyzing the Runx1b-expressing subaortic mesenchymal cells, Fadlullah et al identified 2 populations representing smooth muscle cells and Pdgfra-expressing mesenchyme. Moreover, Runx1b deletion resulted in an increase in smooth muscle cells, accompanied by the enrichment of genes involved in ribosomal transcripts and proliferation.
Taking advantage of the high sensitivity of the data set, Fadlullah et al further screened and identified Ace as a new surface marker with higher expression in pre-HE than in other adjacent populations; it had not been robustly detected in prior studies.5,8 Interestingly, pre-HE was mainly derived from Runx1b reporter–positive cells but seldom expressed Runx1 transcripts. The situation is believed to result from the compromised ability to detect low levels of Runx1 transcripts, even using a full-length sequencing method. The possible inconsistency between transcriptional and phenotypic (protein or reporter) levels for a given gene emphasizes the requirement of additional verification for important candidates predicted by the scRNA-seq data. Ace was expressed specifically in the aortic endothelial layer but not in other vasculatures nor in intra-aortic hematopoietic clusters. Functionally, all induced hematopoietic potential in nonhematopoietic cells was confined to Ace-positive endothelium. These findings collectively indicate that Ace marks the whole HE continuum. Based on previous identification of pre-HE by transcriptomic characteristics and regulatory elements,5 the present findings improve our understanding of this endothelial population lying upstream of HE. The next step could be to decipher the physiological fate of pre-HE marked by Ace expression, using potential lineage-tracing models. Considering the remarkable arterial characteristics therein, the extent to which it contributes to adult HSCs and aortic vasculature will be interesting.
Taken together, this set of scRNA-seq data provide unprecedented multi-layer resources for future studies of HSC occurrence, covering microenvironment cells and cells lacking key transcription factors, as well as phenotypic correspondence information and isoform-level expression profiles of the sequenced populations. Because previous studies have found that the expression of genes involved in the regulation of RNA splicing changes with EHT process,4,7 future exploration of the molecular mechanisms at the isoform level with the help of this full-length scRNA-seq dataset is anticipated. The integration of this data set with the corresponding human data is also desirable.9 Evolutionary conservation analysis, including the comparison of the subaortic niche between the species, would help us to better understand the emergence of mammalian HSCs and provide theoretical support for regenerating clinically available HSCs.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal