

Key Points
SAR442085 is a novel Fc-engineered anti-CD38 monoclonal antibody with enhanced affinity for FcγRIIa and FcγRIIIa receptors.
SAR442085 has higher NK cell-dependent in vitro and in vivo antimyeloma efficacy than standard-of-care daratumumab and isatuximab.

Abstract
Anti-CD38 monoclonal antibodies (mAbs) represent a breakthrough in the treatment of multiple myeloma (MM), yet some patients fail to respond or progress quickly with this therapy, highlighting the need for novel approaches. In this study we compared the preclinical efficacy of SAR442085, a next-generation anti-CD38 mAb with enhanced affinity for activating Fcγ receptors (FcγR), with first-generation anti-CD38 mAb daratumumab and isatuximab. In surface plasmon resonance and cellular binding assays, we found that SAR442085 had higher binding affinity than daratumumab and isatuximab for FcγRIIa (CD32a) and FcγRIIIa (CD16a). SAR442085 also exhibited better in vitro antibody-dependent cellular cytotoxicity (ADCC) against a panel of MM cells expressing variable CD38 receptor densities including MM patients’ primary plasma cells. The enhanced ADCC of SAR442085 was confirmed using NK-92 cells bearing low and high affinity FcγRIIIa (CD16a)-158F/V variants. Using MM patients’ primary bone marrow cells, we confirmed that SAR442085 had an increased ability to engage FcγRIIIa, resulting in higher natural killer (NK) cell activation and degranulation against primary plasma cells than preexisting Fc wild-type anti-CD38 mAbs. Finally, using huFcgR transgenic mice that express human Fcγ receptors under the control of their human regulatory elements, we demonstrated that SAR442085 had higher NK cell-dependent in vivo antitumor efficacy and better survival than daratumumab and isatuximab against EL4 thymoma or VK*MYC myeloma cells overexpressing human CD38. These results highlight the preclinical efficacy of SAR442085 and support the current evaluation of this next-generation anti-CD38 antibody in phase I clinical development in patients with relapsed/refractory MM.
Introduction
Multiple myeloma (MM) is a devastating hematological cancer characterized by the proliferation of clonal, long-lived plasma cells (PC) within the bone marrow (BM) and is associated with bone destruction, serum monoclonal gammopathy (M-spike), and organ dysfunctions.1 Although drugs such as proteasome inhibitors and immune modulatory drugs (IMiDs) have resulted in remarkable survival improvement in the last 10 years, most patients experience recurrent relapses with shorter periods of remissions and have limited therapeutic options. The introduction of monoclonal antibodies (mAbs) targeting CD38, an ectoenzyme highly expressed at the membrane of malignant PCs, has represented a major breakthrough in the treatment of MM.2-6 The anti-CD38 mAb daratumumab (Darzalex, Janssen of Johnson & Johnson) was the first to be approved both in relapsed/refractory and newly diagnosed MM. More recently, isatuximab (Sarclisa), targeting a CD38 epitope different from daratumumab, was also approved in combination therapy in relapsed/refractory MM.7-9
CD38-targeting antibodies rely on several mechanisms of action to exert their antimyeloma activity.10-13 Accumulating evidence points toward the importance of Fc-dependent immune-effector mechanisms for the antimyeloma efficacy of these drugs.14-16 Indeed, anti-CD38 mAbs were shown to induce the killing of malignant PCs through antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and complement-dependent cytotoxicity (CDC).12,13 Of note, ADCP and ADCC rely on the interaction of the antibody Fc domain with distinct activating Fc-γ receptors (FcγR): FcγRIIIa (CD16a) and FcγRIIa (CD32a) on monocytes and macrophages for ADCP and FcγRIIIa on natural killer (NK) cells for ADCC. In addition, Fc-dependent depletion of CD38+ immunosuppressive cells such as myeloid-derived suppressor cells and Tregs may also contribute to the antimyeloma activity of anti-CD38 mAbs by increasing T cell reactivity against myeloma cells.10,17-20 Direct proapoptotic activity and CD38 ectoenzyme inhibition were also shown to contribute to the antitumor activity of isatuximab,10,14,21,22 and to a lesser extent, of daratumumab, for which apoptosis was revealed only upon FcR-mediated crosslinking.15,23
Despite the clinical benefits of daratumumab in newly diagnosed and relapsed/refractory MM, primary resistance and early progressions can be observed, stressing the need for novel treatments.24,25 The importance of immune-dependent mechanisms in the efficacy of anti-CD38 mAbs suggests that increasing their affinity toward activating FcγR may represent a promising therapeutic strategy to improve their clinical efficacy,26 especially given the existence of FCGR2A and FCGR3A polymorphisms leading to FcγRIIa (131 H/R) and FcγRIIIa (158 F/V) allelic variants with different IgG Fc domain binding affinity.27-29 The high prevalence of FcγRIIa-131H and FcγRIIIa-158F low affinity variants in the population reinforces the importance of engineering Fc-enhanced antibodies that can bind to both receptor variants with increased affinity and thus overcome the Fc-FcγR interaction shortfalls.27 Previous Fc-engineered mAbs either by afucosylation (glyco-engineered Fc)30 or by introducing point mutations into the Fc domain have shown superior activity without increased toxicity compared with their nonengineered counterparts. ADCC-enhanced mAbs, such as obinutuzumab (Gazyva) anti-CD20 mAb and, more recently, margetuximab anti-HER2 mAb, have been approved, and many others are in clinical development for cancer indications.26,31-33
In this study, we compared the preclinical efficacy of SAR442085, a next-generation human Fc-engineered IgG1 backbone anti-CD38 antibody, with the first generation and most frequently used anti-CD38 antibodies daratumumab and isatuximab. Our results revealed the increased affinity of SAR442085 for the low and high affinity variants of both FcγRIIIa and FcγRIIa, resulting in enhanced in vitro antimyeloma activity of NK cells and macrophages compared with daratumumab and isatuximab regardless of FCGR2A and FCGR3A polymorphism. The enhanced NK cell-dependent ADCC of SAR442085 was confirmed in vitro against MM cell lines expressing low CD38 antigen density and against primary MM patients' tumor cells and in mouse tumor models mimicking MM pathology. Altogether, this study reveals the promising therapeutic potential of SAR442085, a novel anti-CD38 Fc engineered antibody.
Methods
Monoclonal antibodies
Clinical-grade daratumumab was obtained from BAP Pharma (Berkshire, UK). Isatuximab was obtained from Sanofi. SAR442085 is an anti-CD38 antibody with humanized variable regions and a human Fc-engineered IgG1 backbone with enhanced FcγR binding (American Association for Cancer Research Annual Meeting 2020, Abstract 2266). SAR442085 was produced in the CHO 9E4 cell line (Sanofi, WO2013186371A1) and prepared and stored in Dulbecco's phosphate-buffered saline (ThermoFisher Scientific). An IgG1-Fc engineered isotype control (named control isotype) was generated with the same Fc modifications and by mutating the variable domain of SAR442085, leading to loss of binding to CD38.
Human samples
Blood samples from healthy donors (HD) were acquired from the “Etablissement Français du Sang” as buffy coats. Fresh BM aspirates and peripheral blood from patients with MM were collected at the time of diagnosis or relapse in the “Institut Universitaire du Cancer de Toulouse-Oncopole” (IUCT-O, Toulouse). All cancer patients gave written informed consent, and collection was approved by French Committee for the Protection of Persons (DC-2012-1654) as well as by local IUCT-O review boards.
Immune and tumor cell isolation
Peripheral blood mononuclear cells (PBMCs) were isolated with Ficoll-Paque Plus (Cytiva) and SepMate tubes (StemCell Technologies) according to the manufacturer instructions. NK cells were purified by negative selection (depletion of T cells, B cells, stem cells, dendritic cells, monocytes, granulocytes, and erythroid cells) from total PBMCs with the Human NK cell isolation kit on the autoMACS Pro Separator (Miltenyi Biotec) according to manufacturer’s instructions. NK cell isolation from the bone marrow of MM patients was performed using NK cell isolation kit in combination with the CD15 microbeads kit (Miltenyi Biotec), which was used to eliminate CD15+ granulocytes present in the BM. Isolated NK cells were then incubated at a concentration of 106 per mL in the presence of hIL-2 at a final concentration of 50 ng/mL overnight at 37°C. Monocytes were isolated from total PBMCs by positive selection with anti-CD14 magnetic beads on the autoMACS Pro Separator. Macrophages were prepared by culturing monocytes for 5 days in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 2% human inactivated serum, and 50 ng/mL granulocyte-macrophage colony-stimulating factor. Malignant plasma cells isolation from MM patients' BM aspirates was performed with the CD138 microbeads kit (Miltenyi Biotec) according to manufacturer's instructions on the autoMACS Pro Separator. The cells were washed, counted, and directly used in functional experiments.
Cell lines
MM cell lines MOLP-8 (ACC 569) and KMS-12-BM (ACC 551) were obtained from the German Collection of Microorganisms and Cell Cultures GmbH (DSMZ), and RPMI-8226 (CCL-155) from the American Type Culture Collection (ATCC). The SU-DHL-8 cell line derived from a B-cell lymphoma was obtained from the German Collection of Microorganisms and Cell Cultures (ACC 573). Cell lines were cultured according to the standard protocol recommendations. The expression of CD38 on MM cells was evaluated by flow cytometry, and CD38 receptor density was calculated using calibrated beads (Cellquant Calibrator, Biocytex). For cellular binding assays, HEK293T cells were genetically engineered to stably express the following Fcγ receptors: FcγRI, FcγRIIa-131H, FcγRIIa-131R, FcγRIIIa-158F, and FcγRIIIa-158V. FcγR-overexpressing cells were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% FBS, 1% l-glutamine, 10 µg/mL blasticidin, and 1 μg/mL puromycin. NK-92.FcγRIIIa-158V (NK-92 modified to express FcγRIIIa high affinity, CD16-158V) and NK-92.FcγRIIIa-158F (NK-92 cells modified to express FcγRIIIa low affinity, CD16-158F) were obtained under license agreement with Conkwest Incorporated. NK-92.FcγRIIIa-158V and NK-92.FcγRIIIa-158F cells were cultured in MyeloCult H5100 medium (StemCell Technologies) supplemented with 40 ng/mL IL-2 (R&D Systems).
Cellular binding assay
The apparent binding affinities to CD38 and FcγR allelic variants were calculated from binding curves obtained by flow cytometry on CD38+ MOLP-8 cells and FcγR-overexpressing HEK293T cells, respectively. A total of 105 cells per sample were incubated in 50 µL phosphate-buffered saline (PBS) with increasing concentrations of SAR442085, daratumumab, and isatuximab (FcγR-overexpressing HEK293T cells) for 30 minutes at 4°C. Cells were then washed three times in PBS supplemented with 1% FBS and stained with fluorescein isothiocyanate (FITC)-conjugated goat anti-human IgG (Jackson ImmunoResearch Laboratories) secondary antibodies at a final concentration of 10 μg/mL. Binding was measured on a MACSQuant VYB flow cytometer (Miltenyi Biotec). Curve fitting and apparent equilibrium dissociation constant (KD) estimations were performed with GraphPad Prism software version 7 using the one-site specific binding model.
Surface plasmon resonance
Binding of SAR442085, isatuximab, and daratumumab to FcγR allelic variants was analyzed by surface plasmon resonance (SPR) using Biacore T200 (Cytiva). His-tagged human FcγRIIIA-158F and FcγRIIIA-158V proteins were captured on anti-His antibodies previously immobilized on CM5 sensorchip by amine coupling according to manufacturer’s instructions. SAR442085, isatuximab, and daratumumab were injected in a single cycle kinetics mode (final concentrations ranging from 0 to 5 µM). For the binding of SAR442085, isatuximab, and daratumumab to CD38, anti-human Fc antibodies were immobilized on CM5 sensorchip according to manufacturer’s instructions allowing to capture each anti-CD38 antibody by its Fc region. CD38 protein produced internally was then injected in a single cycle kinetics mode (final concentrations ranging from 0 to 100 nM). The sensorgrams were fitted with Langmuir 1:1 model using the Biacore Evaluation Software 3.1.
Antibody-dependent cellular cytotoxicity
ADCC was measured by the calcein release method according to manufacturer’s instructions. Briefly, MM cells were stained with calcein AM (Invitrogen) at 37°C for 30 minutes. Following three washing steps, cells were plated on a round-bottom 96-well plate (at a density of 20 000 cells per well) and incubated with SAR442085, isatuximab, or daratumumab for 30 minutes at 37°C. NK-92 effector cells were then added at a density of 100 000 cells per well (effector to target cells ratio of 5:1). Following a 1-hour incubation at 37°C, calcein released in the supernatant by dying tumor cells was measured using a fluorescence plate reader (Infinite M1000 PRO, TECAN). The specific lysis was calculated relative to a positive control consisting of tumor cells cultured in medium supplemented with 2% Triton X-100 (Sigma-Aldrich). Alternatively, ADCC was evaluated overnight by flow cytometry with propidium iodide (PI). MM cells were labeled with CellTrace Violet (CTV) at 37°C for 20 minutes. Target cells (2 × 104 cells per well) were then preincubated with antibodies for 30 minutes at 37°C before the addition of effector cells at an effector to target ratio of 50:1 (PBMCs) or 10:1 (purified NK cells). ADCC with PBMCs from HDs was performed in the presence of 3 mg/mL of human polyclonal IgG (Sigma-Aldrich) to mimic physiological conditions (average immunoglobulin content in the blood of MM patients). The percentage of ADCC was calculated based on the number of PI positive dead cells among CTV positive tumor cells.
Antibody-dependent cellular phagocytosis
ADCP was evaluated by preincubating 5 × 104 PKH67 (Sigma-Aldrich)-labeled RPMI-8226 target cells for 15 minutes in round-bottom 96-well plates with SAR442085, isatuximab, or daratumumab. Human monocyte-derived macrophages labeled with PKH26 (Sigma-Aldrich) were then added (effector to target cells ratio of 3:1) and incubated overnight with tumor cells and antibodies at 37°C. ADCP assay was performed in the presence of 1 mg/mL human polyclonal IgG (Sigma-Aldrich). Phagocytosis was calculated as the percentage of PKH26+PKH67+ cells relative to all PKH67+ cells detected by flow cytometry.
CD38 ecto-enzymatic activity
CD38 enzyme inhibition was analyzed with RPMI8226 cells that were incubated with 20 µg/mL of isotope labeled nicotinamide adenosine diphosphate (N15-NAD) as a substrate for CD3834 and SAR442085, daratumumab, isatuximab, or isotype control. After 1 hour at 37°C, the cells culture supernatant was transferred to a 96-well plate, and the amount of NAD converted to cyclic adenosine 5′-diphosphate (ADP)-ribose (cADPR) was measured by LC/MS/MS on a Sciex Qtrap 5500 Mass spectrometer coupled with Agilent Infinity 1290 UHPLC system. The peak area of cADPR on the chromatogram was integrated using Sciex Analyst software and plotted vs the antibody concentration for a dose response experiment.
Apoptosis induction
SU-DHL-8 cells, RPMI-8226, or MOLP-8 (1 × 105 cells per well) were incubated with anti-CD38 antibody in 96-well flat-bottom plates for 24 hours at 37°C. The induction of apoptosis was measured by flow cytometry with Annexin V-FITC and PI staining kit (Miltenyi Biotec) in accordance with manufacturer's instructions.
Complement-dependent cytotoxicity
CDC activity of anti-CD38 antibodies was measured with a nonradioactive method based on the method described by Gazzano-Santoro et al.35 Briefly, SU-DHL-8 cells were plated in a 96-well black/clear flat-bottom tissue culture plate (1 × 105 cells per well) in ice-cold RHB medium (RPMI 1640 medium, no phenol red supplemented with 2 mM L-Glutamine, 2 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid and 0.1% bovine serum albumin) and then incubated on ice for 1 hour with each antibody. Human complement was added to each well and the plate incubated for 4 hours at 37°C. Alamar Blue reagent (ThermoFisher Scientific) was added to each well and the fluorescence was measured 4 hours later with a fluorescence plate reader according to manufacturer’s instructions.
Analysis of NK cell functions
MM patient PC or MM cell lines were incubated for 30 minutes at 37°C with the different anti-CD38 mAbs (SAR442085, isatuximab, and daratumumab) at the following concentrations: 0.07, 0.7, 7, 70, and 700 pM. An IgG control antibody containing SAR442085 variable regions with mutated binding site was used as a negative control at the maximal concentration of 100 ng/mL. Cells were washed twice with ice-cold PBS before being cocultured with purified NK cells from HD or MM patients at a ratio of 1:10 (target:NK cell). NK cell degranulation, CD69, and FcγRIIIa expression were analyzed after 4 hours at 37°C in the presence of APC-conjugated anti-CD107a (H4A3, BD Biosciences). Cytokine levels were measured in the cell culture supernatants by Cytometric Bead Array kit (BD Biosciences) according to manufacturer’s instructions and analyzed using FCAP array V3 (BD Biosciences).
Flow cytometry
Single cell suspensions were stained according to standard protocols with previously described anti-mouse and human antibodies. Antibodies were purchased from Miltenyi Biotec, eBioScience, BioLegend, or BD Biosciences. Surface staining was performed with antibodies for 30 minutes at 4°C in PBS supplemented with 2% FCS and 2 mM EDTA. For the intracellular staining, cells were fixed and permeabilized using BD Cytofix/Cytoperm permeabilization kit (BD Biosciences) or Transcription Factor Fixation/Permeabilization kit (Thermo Fisher Scientific). The antibodies used in this study were: BUV737 mouse anti-human CD56 (Clone NCAM16.2; BD Biosciences; Ref564447; 1/200); FITC anti-human CD38 (Clone HB-7; Biolegend; Ref356610; 1/100); FITC mouse anti-human CD138 (Clone Ml15; BD Biosciences; Ref561703; 1/100); PerCP-Cy5.5 mouse anti-human FcγRIIIa (Clone 3G8; BD Biosciences; Ref560717; 1/100); BV711 mouse anti-human CD69 (Clone FN50; BD Biosciences; Ref563836; 1/100); APC mouse anti-human CD107a (Clone H4A3; BD Biosciences; 641581); PE-cy7 mouse anti-human CD38 (Clone HB-7; BD Biosciences); and BV421 rat anti-mouse CD138; PE Rat anti-mouse CD155. Data were collected with LSR II or Fortessa X20 flow cytometers (BD Biosciences) and analyzed with FlowJo software (TreeStar). Dead cells and doublets were excluded by LiveDead staining (Thermo Fisher Scientific).
Mouse models
All mice used in these studies were on a C57BL/6 genetic background. Wild-type mice were purchased from Janvier laboratories. Rag2−/−Il2rg−/−36 have already been described. The immune competent mice expressing only the human FcγRs used for in vivo experiments were created and obtained by Jeffrey V. Ravetch laboratory37 from Rockefeller University, USA. This mouse model contains each of the individual huFcγR transgenes (huFcγRI, huFcγRIIAR131, huFcγRIIBI232, huFcγRIIIAF158, and huFcγRIIIB) under the control of their human promoters and regulatory elements. Male and female huFcγR transgenic (tg) C57BL/6 mice were bred in specific and opportunistic pathogen-free conditions at the animal facility of “Centre de Recherche et Développement de Montpellier” (Sanofi).
EL4-huCD38 disseminated model
The murine T-cell lymphoma, EL4 cell line (TIB-39) was obtained from the American Type Culture Collection. EL4 murine T lymphoma cells were infected by a nonreplicative lentivirus with human CD38 (EL4-huCD38). EL4-huCD38 cells were cultured under blasticidin selection to maintain optimal level of sequence integration. huFcγR transgenic C57BL/6 mice were housed in specific and opportunistic pathogen-free conditions at the animal facility of Centre de Recherche de Vitry-Alfortville (Sanofi). All animal procedures were approved by the Sanofi Animal Care and Use Committee and followed the French and European regulations on care and protection of the laboratory animals. The disseminated tumor model was established in huFcγR mice by injecting IV 5 × 105 cells per mouse in PBS suspension in the tail vein at day 0. Mice were subsequently treated by intraperitonal (IP) injection of SAR442085, isatuximab, daratumumab, or isotype control (10 mg/kg and 1.25 mg/kg) on days 1, 4, 7, 11, and 14. For survival analysis, mice were monitored daily according to institutional ethic guidelines and were killed when mice developed signs of reduced mobility including paralysis, hunched posture, or respiratory distress. Splenocytes from long-term surviving mice (SAR442085, isatuximab, and daratumumab groups), killed isotype-treated mice, and naïve huFcγR mice were stained according to standard protocol. The following antibodies were used to identify immune cells population: anti-F4/80 Alexa Fluor 700 (BM8), anti-NK1.1 BV605 (PK136), anti-CD3 BV650 (17A2), anti-CD11c BV786 (N418) from Biolegend, anti-Ly6C/6G APC Alexa 750 (RB6-8C5), anti-CD11b PE Texas Red (M1/70.15), LIVE/DEAD Fixable Aqua Dead Cell Stain from Thermo Fisher Scientific, and CD45 PECY7 (30-F11) from BD Biosciences.
HuCD38 VK*MYC model
Transplantable VK*MYC MM cell line (Vk12653) was kindly provided by L. Bergsagel and M. Chesi (Mayo Clinic, Rochester). Vk12653 cells were expanded as previously described.38,39 Briefly, these cells were maintained in Rag2−/−Il2rg−/− mice to avoid contamination with host-derived lymphocytes as described previously.39 Mice were housed in the specific pathogen-free animal facility of the US006 CREFRE-Inserm/UPS, which is accredited by the French Ministry of Agriculture (accreditation number A-31 55508). Tumor experiments used both male and female mice between 6 and 12 weeks of age. Donors and recipients of adoptive NK cell transfers were sex-matched. Animal experiments were conducted and approved by the Ministère de l'Enseignement Supérieur, de la Recherche et de l'Innovation (APAFIS#5614-20 16060815487810 v4) and following the French and European regulations on care and protection of the laboratory animals. To generate Vk12653-huCD38 MM cells, highly concentrated human CD38 integrative lentiviral vectors (Flash Therapeutics) were used. Briefly, splenocytes from Vk12653 bearing Rag2−/−Il2rg−/− mice were harvested and transduced in vitro with huCD38 lentiviral vector at a multiplicity of infection of 10 on retronectin-coated plates for 1 hour. As VK*MYC cells do not survive in culture, transduced cells were quickly reinjected IV into Rag2−/−Il2rg−/−mice. After 5 weeks, CD38 expressing malignant PCs were sorted from the spleen of tumor-bearing mice and reinjected into new Rag2−/−Il2rg−/− mice. Three rounds of sorting and reinjection of CD38+ CD138+CD155+ cells were performed to stably generate >90% of CD38-expressing Vk12653 cells. Splenocytes containing >50% of malignant PCs expressing human CD38 at >90% were kept frozen until use.
In vivo NK cell ADCC against HuCD38 VK*MYC model
0.2x106 NK cells (NK1.1+ CD3-) were isolated using fluorescence-activated cell sorting (FACS) from the spleen of huFcγR mice and injected IV into the tail vein of Rag2−/−Il2rg−/− mice. One week later, NK cell reconstitution was monitored by eye bleeding, and MM was induced by IV injection of Vk12653-huCD38 cells. Mice were treated by IP injection of SAR442085, isatuximab, daratumumab, or isotype control (10 mg/kg; 6 injections every 2-3 days) starting at day 7 relative to tumor injection. The percentage of monoclonal Ig in the serum was quantified weekly by serum protein electrophoresis (Sebia Hydrasys system) to evaluate differences in myeloma growth between groups.
Relative EC50 calculation and statistical analysis
Binding assays
Curve fitting and Kd estimations were performed using the one-site specific binding model for each experiment. For KD, statistical differences between groups (SAR442085 vs daratumumab or isatuximab) were determined after log transformation by an ordinary one-way analysis of variance (ANOVA) (KD) or a repeated measure one-way ANOVA (apparent KD) followed by Dunnett's multiple comparisons test. For Bmax, statistical differences between groups were determined by a repeated measure one-way ANOVA followed Dunnett's multiple comparisons test.
ADCC and ADCP assays
A dose-response curve was fitted, and the relative half-maximal effective concentration (EC50rel) was determined for each experiment using the 4-parameter logistic model according to Ratkowsky and Reedy.40 The Geometric means and 95% confidence interval of EC50rel were calculated for each compound. For EC50rel, statistical differences between groups (SAR442085 vs daratumumab or isatuximab) were determined after log transformation by a repeated measure one-way ANOVA followed by Dunnett's multiple comparisons test. For maximum killing, statistical differences between groups were determined by a repeated measure one-way ANOVA followed by Dunnett's multiple comparisons test. For graphical representations of binding and ADCC/ADCP assays, one curve was fitted for all experiments (with mean of replicates by donor).
Percentages of the splenic immune cell populations and for M spike
Statistical differences between groups of mice were performed using Dunn’s tests after a Kruskal-Wallis test.
Mouse models assays
The survival of mice and the evolution of paraproteinemia-free mice were represented by Kaplan-Meier curves and were compared between groups using log-rank tests followed by a Holm-Sidak correction for multiplicity.
Evaluation of the NK cell functions on MM patients’ PC or MM cell lines
For each parameter, statistical differences between groups were determined by a two-way ANOVA with Bonferroni-Holm correction (for IFN-γ and MIP1-α cytokines, this analysis was performed on squared root transformed data).
All these statistical analyses were performed using GraphPad Prism Software version 7 or 8 or SAS version 9.4. P < .05 was considered statistically significant (P < .05 = *; P < .01 = **; P < .001 = ***).
Results
Fc-engineered anti-CD38 mAb, SAR442085, exhibits superior binding affinity to FcγRIIIa and FcγRIIa receptors than daratumumab
A humanized IgG1 anti-CD38 mAb, SAR442085 (American Association for Cancer Research Annual Meeting 2020, Abstract 2266), engineered in the Fc domain in order to increase its binding to Fcγ receptors was recently generated. We first analyzed the CD38 binding affinity of SAR442085 by SPR using recombinant human CD38 and by flow cytometry using CD38-positive MOLP-8 MM cells and found dissociation constants (KD) and apparent KD of 0.2 nM and 1.1 nM, respectively (supplemental Figure 1A-B; Table 1). Of note, the current anti-CD38 mAb reference in myeloma, daratumumab, had a slightly lower affinity for CD38 with Kd (SPR) and apparent Kd (flow cytometry) of 1.4 nM and 1.9 nM, respectively (supplemental Figure 1A-B; Table 1). FcγRIIIa (V158F) being associated with differential affinity toward mAbs,28 we then compared the binding of SAR442085, daratumumab, and isatuximab to FcγRIIIa-158F (low affinity) and 158V (high affinity) variants by SPR. With a Kd of 119 nM for FcγRIIIa-158F and 46 nM for FcγRIIIa-158V, SAR442085 had a 21- and sevenfold higher binding affinity to FcγRIII-158F and 158V, respectively, compared with daratumumab (Figure 1A-B; Table 2). The enhanced affinity of SAR442085 compared with daratumumab and isatuximab for FcγRIIIa low and high affinity allelic variants was confirmed by flow cytometry using FcγRIIIa-158F or 158V-overexpressing HEK293T cells (Figure 1C-D; supplemental Table 1).
Cytotoxic activity of SAR442085, daratumumab and isatuximab against the indicated MM cell lines in the presence of NK92-FcγRIIIa cells
| Target cells | Effector cells | Compound | Geometric mean EC50rel (pM) [95% CI]* | Fold improvement† |
|---|---|---|---|---|
| RPMI 8226 | NK92-FcγRIIIa158F | SAR442085 | 3.121 [0.19; 51.44] | 24 |
| Daratumumab | 73.26 [6.25; 858.3] | - | ||
| Isatuximab | 84.59 [2.11; 3398] | - | ||
| NK92-FcγRIIIa158V | SAR442085 | 8.41 [0.75; 94.54] | 14 | |
| Daratumumab | 118.8 [9.5; 1486] | - | ||
| Isatuximab | 65.74 [4.26; 1014] | |||
| MOLP8 | NK92-FcγRIIIa158F | SAR442085 | 1.28 [0.20; 8.39] | 28 |
| Daratumumab | 36.07 [10.68; 121.8] | - | ||
| Isatuximab | 30.28 [7.3; 125.6] | |||
| NK92-FcγRIIIa158V | SAR442085 | 2.82 [0.24; 33.18] | 15 | |
| Daratumumab | 42.21 [6.21; 287] | - | ||
| Isatuximab | 32.8 [4.69; 230] | |||
| KMS-12-BM | NK92-FcγRIIIa158F | SAR442085 | 1.969 [0.103; 37.75] | 22 |
| Daratumumab | 43.29 [1.29; 1448] | - | ||
| Isatuximab | 53.5 [2.77; 1034] | |||
| NK92-FcγRIIIa158V | SAR442085 | 4.65 [0.97; 22.39] | 13 | |
| Daratumumab | 58.58 [15.15; 310.5] | - | ||
| Isatuximab | 65.74 [4.26; 1014] |
| Target cells | Effector cells | Compound | Geometric mean EC50rel (pM) [95% CI]* | Fold improvement† |
|---|---|---|---|---|
| RPMI 8226 | NK92-FcγRIIIa158F | SAR442085 | 3.121 [0.19; 51.44] | 24 |
| Daratumumab | 73.26 [6.25; 858.3] | - | ||
| Isatuximab | 84.59 [2.11; 3398] | - | ||
| NK92-FcγRIIIa158V | SAR442085 | 8.41 [0.75; 94.54] | 14 | |
| Daratumumab | 118.8 [9.5; 1486] | - | ||
| Isatuximab | 65.74 [4.26; 1014] | |||
| MOLP8 | NK92-FcγRIIIa158F | SAR442085 | 1.28 [0.20; 8.39] | 28 |
| Daratumumab | 36.07 [10.68; 121.8] | - | ||
| Isatuximab | 30.28 [7.3; 125.6] | |||
| NK92-FcγRIIIa158V | SAR442085 | 2.82 [0.24; 33.18] | 15 | |
| Daratumumab | 42.21 [6.21; 287] | - | ||
| Isatuximab | 32.8 [4.69; 230] | |||
| KMS-12-BM | NK92-FcγRIIIa158F | SAR442085 | 1.969 [0.103; 37.75] | 22 |
| Daratumumab | 43.29 [1.29; 1448] | - | ||
| Isatuximab | 53.5 [2.77; 1034] | |||
| NK92-FcγRIIIa158V | SAR442085 | 4.65 [0.97; 22.39] | 13 | |
| Daratumumab | 58.58 [15.15; 310.5] | - | ||
| Isatuximab | 65.74 [4.26; 1014] |
EC50rel, relative EC50; 95% CI, 95% confidence interval.
EC50rel fold improvement of SAR442085 vs. daratumumab.
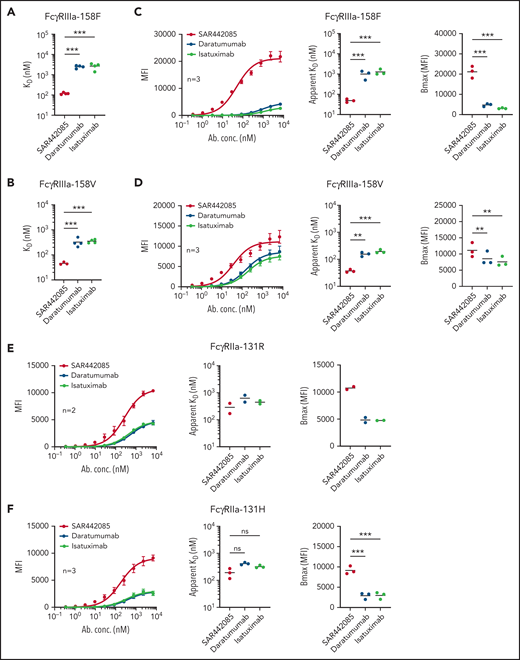
SAR442085 has a higher binding affinity for activating FcγRIIa and FcγRIIIa than daratumumab and isatuximab. (A-B) Binding of SAR442085, daratumumab, or isatuximab to FcγRIIIa-158V and FcγRIIIa-158F was evaluated by SPR using purified human proteins. Graphs represent the mean and individual KD data. Statistical differences between groups were determined by an unpaired Student t-test. (C-F) Cellular binding of SAR442085, daratumumab, or isatuximab to FcγRIIa and FcγRIIIa receptor variants was evaluated by flow cytometry on HEK293T cells overexpressing each variant. Graphs represent the pooled binding curves of up to 3 independent experiments with the mean and individual values of apparent Kd and maximum binding (Bmax) for each binding curve. MFI, mean fluorescence intensity; KD, dissociation constant; Bmax, maximal amount of antibody bound at the cell surface. Statistical differences between groups were determined by a ratio paired t test. *P < .05; **P < .01, ***P < .001.
SAR442085 has a higher binding affinity for activating FcγRIIa and FcγRIIIa than daratumumab and isatuximab. (A-B) Binding of SAR442085, daratumumab, or isatuximab to FcγRIIIa-158V and FcγRIIIa-158F was evaluated by SPR using purified human proteins. Graphs represent the mean and individual KD data. Statistical differences between groups were determined by an unpaired Student t-test. (C-F) Cellular binding of SAR442085, daratumumab, or isatuximab to FcγRIIa and FcγRIIIa receptor variants was evaluated by flow cytometry on HEK293T cells overexpressing each variant. Graphs represent the pooled binding curves of up to 3 independent experiments with the mean and individual values of apparent Kd and maximum binding (Bmax) for each binding curve. MFI, mean fluorescence intensity; KD, dissociation constant; Bmax, maximal amount of antibody bound at the cell surface. Statistical differences between groups were determined by a ratio paired t test. *P < .05; **P < .01, ***P < .001.
Cytotoxic activity of SAR442085, daratumumab and isatuximab against the indicated MM cell lines in the presence of total PBMCs, purified NK cells, and monocyte-derived macrophages from healthy donors
| Target cells | Effector cells | Compound | Geometric mean EC50rel (pM) [95% CI]* | Fold improvement† |
|---|---|---|---|---|
| RPMI 8226 | HD PBMC | SAR442085 | 54.63 [35.7 ; 83.7] | 3 |
| Daratumumab | 182.9 [76.82 ; 735.6] | |||
| Isatuximab | 130.8 [72.37 ; 236.3] | |||
| KMS-12-BM | HD PBMC | SAR442085 | 14.78 [7.33 ; 29.79] | 5 |
| Daratumumab | 73.44 [36.9 ; 146.1] | |||
| Isatuximab | 69.2 [32.79 ; 146.4] | |||
| RPMI 8226 | HD NK | SAR442085 | 1.07 [0.08 ; 14.49] | 10 |
| Daratumumab | 10.83 [5.68 ; 20.64] | |||
| Isatuximab | 11.64 [4.11 ; 32.98] | |||
| MOLP8 | HD monocyte-derived macrophages | SAR442085 | 18.35 [12.72 ; 26.45] | 4 |
| Daratumumab | 66.33 [20.6 ; 213.3] | |||
| Isatuximab | 55.51 [17.38 ; 177.3] |
| Target cells | Effector cells | Compound | Geometric mean EC50rel (pM) [95% CI]* | Fold improvement† |
|---|---|---|---|---|
| RPMI 8226 | HD PBMC | SAR442085 | 54.63 [35.7 ; 83.7] | 3 |
| Daratumumab | 182.9 [76.82 ; 735.6] | |||
| Isatuximab | 130.8 [72.37 ; 236.3] | |||
| KMS-12-BM | HD PBMC | SAR442085 | 14.78 [7.33 ; 29.79] | 5 |
| Daratumumab | 73.44 [36.9 ; 146.1] | |||
| Isatuximab | 69.2 [32.79 ; 146.4] | |||
| RPMI 8226 | HD NK | SAR442085 | 1.07 [0.08 ; 14.49] | 10 |
| Daratumumab | 10.83 [5.68 ; 20.64] | |||
| Isatuximab | 11.64 [4.11 ; 32.98] | |||
| MOLP8 | HD monocyte-derived macrophages | SAR442085 | 18.35 [12.72 ; 26.45] | 4 |
| Daratumumab | 66.33 [20.6 ; 213.3] | |||
| Isatuximab | 55.51 [17.38 ; 177.3] |
EC50rel: Relative EC50. 95% CI: 95% confidence interval.
EC50rel fold improvement of SAR442085 vs. daratumumab.
Similarly, FCGR2A polymorphism has been proposed to impact the therapeutic efficacy of tumor targeting antibodies.28 Accordingly, the binding of SAR442085, daratumumab, and isatuximab to activating FcγRIIa receptors was evaluated by flow cytometry using HEK293T cells overexpressing FcγRIIa-131R or FcγRIIa-131H allelic variants. Once again, we observed a higher affinity of SAR442085 toward both FcγRIIa-131R and FcγRIIa-131H variants as compared with daratumumab and isatuximab (Figure 1E-F). The binding of SAR442085 to FcγRIIa variants was also evaluated by SPR and showed a KD of 720 nM and 2150 nM for FcγRIIa-131H and -131R, respectively. In this setting, daratumumab and isatuximab appeared weak binders (-131H) or nonbinders (-131R) (supplemental Table 2A-B). Of note, SAR442085 also bound to FcγRIIb receptor with a Kd of 396 nM and FcγRI receptor with a Kd of 20 nM. Altogether, these results show that SAR442085 is a novel anti-CD38 antibody with a subnanomolar affinity for CD38 and a higher affinity for the activating FcγRIIIa and FcγRIIa receptor allelic variants as compared with daratumumab and isatuximab.
SAR442085 exhibits superior ADCC and ADCP activities
The mechanism of action of tumor targeting CD38 mAbs typically involves ADCC by NK cells through FcγRIIIa-dependent mechanisms.15,21 Given the increased binding capacity of SAR442085 to FcγRIIIa receptors, we compared the ADCC activity of SAR442085, daratumumab, and isatuximab using NK-92 NK cells expressing FcγRIIIa-158V/V or FcγRIIIa-158F/F. As most MM patients display on average between 50 000 and 200 000 CD38 sites per cell,22 RPMI-8226 (approximately 90 000 CD38 receptors per cell) was first selected as a preferential target (supplemental Figure 2A). Both FcγRIIIa-158V and FcγRIIIa-158F NK-92 cells exhibited enhanced ADCC activity against RPMI-8226 in the presence of SAR442085 compared with daratumumab and isatuximab (Figure 2A-B). The half-maximal effective concentration (EC50) of SAR442085 was 14- to 24-fold lower than daratumumab for FcγRIIIa-158V and FcγRIIIa-158F, respectively (Table 1). Similar results were obtained using MOLP-8 (500 000 CD38 molecules per cell, supplemental Figure 2A) and KMS-12-BM (14 000 CD38 molecules per cell, supplemental Figure 2A) that represent CD38 high and CD38 low MM cell lines, respectively (Figure 2C-F; Table 1).
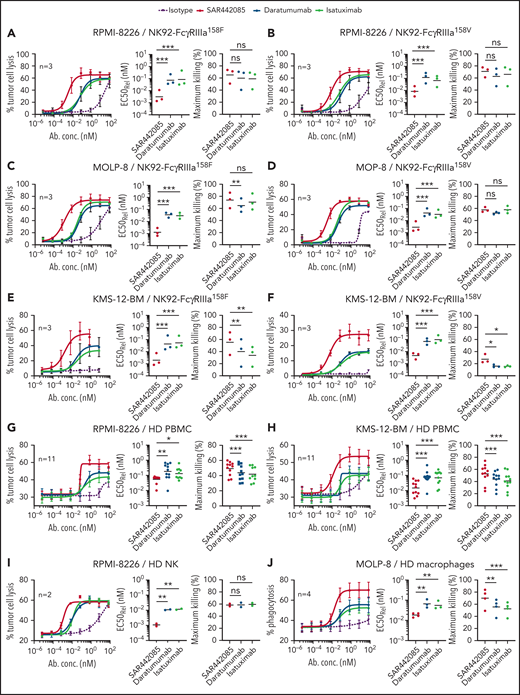
SAR442085 shows enhanced antitumor ADCC and ADCP activity in vitro as compared with daratumumab and isatuximab. (A-F) The ADCC activity of NK-92 cells expressing FcγRIIIa-158F or FcγRIIIa-158V against RPMI-8226 (A-B), MOLP8 cells (C-D), or KMS-12-BM cells (E-F) was analyzed in the presence of increasing concentrations of Ig control, SAR442085, daratumumab, or isatuximab by calcein release assay after 1 hour at an effector to target ratio of 5:1. (G-H) The tumor cell lysis of CTV-labeled RPMI-8226 (G) or KMS-12-BM (H) cell lines incubated with increasing concentrations of Ig control, SAR442085, daratumumab, or isatuximab was evaluated by flow cytometry in the presence of healthy donor PBMCs after 18 hours at an effector to target ratio of 50:1. (I) The ADCC activity against CTV-labeled RPMI-8226 by healthy donor NK cells (HD NK) was measured by calcein release in the presence of the indicated mAbs (effector/tumor ratio = 10:1). (J) The phagocytosis was evaluated by flow cytometry using PKH26-labeled macrophages and PKH67-labeled RPMI8826 cells. All graphs represent the mean ± standard error of the mean (SEM) of pooled dose-response curves. Relative EC50 values (geometric mean) and maximum killing values (mean) for each dose-response curve were calculated. Statistical differences between groups were determined by a ratio paired t test for relative EC50 and by a paired Student t-test for maximum killing values. *P < .05; **P < .01, ***P < .001.
SAR442085 shows enhanced antitumor ADCC and ADCP activity in vitro as compared with daratumumab and isatuximab. (A-F) The ADCC activity of NK-92 cells expressing FcγRIIIa-158F or FcγRIIIa-158V against RPMI-8226 (A-B), MOLP8 cells (C-D), or KMS-12-BM cells (E-F) was analyzed in the presence of increasing concentrations of Ig control, SAR442085, daratumumab, or isatuximab by calcein release assay after 1 hour at an effector to target ratio of 5:1. (G-H) The tumor cell lysis of CTV-labeled RPMI-8226 (G) or KMS-12-BM (H) cell lines incubated with increasing concentrations of Ig control, SAR442085, daratumumab, or isatuximab was evaluated by flow cytometry in the presence of healthy donor PBMCs after 18 hours at an effector to target ratio of 50:1. (I) The ADCC activity against CTV-labeled RPMI-8226 by healthy donor NK cells (HD NK) was measured by calcein release in the presence of the indicated mAbs (effector/tumor ratio = 10:1). (J) The phagocytosis was evaluated by flow cytometry using PKH26-labeled macrophages and PKH67-labeled RPMI8826 cells. All graphs represent the mean ± standard error of the mean (SEM) of pooled dose-response curves. Relative EC50 values (geometric mean) and maximum killing values (mean) for each dose-response curve were calculated. Statistical differences between groups were determined by a ratio paired t test for relative EC50 and by a paired Student t-test for maximum killing values. *P < .05; **P < .01, ***P < .001.
We confirmed the higher killing potency (three to 10-fold improvement) of SAR442085 compared with daratumumab and isatuximab using HD-derived PBMCs as effector cells against RPMI-8226 and KMS-12-BM MM cell lines (Figure 2G-H; Table 2) or using purified HD-derived NK cells against RPMI-8226 cells (Figure 2I; Table 2). Consistent with its higher FcγRIIa and FcγRIIIa affinity, SAR442085 also induced stronger HD monocyte-derived macrophage-mediated ADCP against MM cells than daratumumab in terms of potency (fourfold enhancement) and efficacy of phagocytosis (70% for SAR442085 vs 56% for daratumumab) (Figure 2J; Table 2). In terms of functional activity, the inhibition of CD38 ecto-enzymatic activity was similar between SAR442085 and isatuximab and significantly higher than daratumumab (supplemental Figure 2B). Both SAR442085 and isatuximab showed a strong direct proapoptotic activity (81% apoptosis) against CD38-positive SU-DHL-8 lymphoma cells in the absence of effector immune cells and without the addition of external cross-linking agent (supplemental Figure 2C). By contrast, SAR442085 and isatuximab induced significantly lower apoptosis of MM cell lines MOLP-8 and RPMI-8226 in the absence of effector cells (supplemental Figure 2D). As described previously,15,23 direct apoptosis was very low with daratumumab without the addition of external cross-linking agent (supplemental Figure 2C-D). Finally, SAR442085 had a lower CDC activity against the SU-DHL-8 lymphoma cells in the presence of human complement than daratumumab with an intermediate activity for isatuximab, which is in agreement with previously published data23 (supplemental Figure 2E). Altogether, these data demonstrate the enhanced ADCC and ADCP activities of SAR442085 as compared with daratumumab and isatuximab against different MM cell lines with low and high CD38 expression levels.
SAR442085 has higher antitumor potency in vivo
Next, we wanted to determine whether SAR442085 has a stronger antitumor activity than daratumumab in vivo. Because FcγR gene expression significantly differs between mouse and human, FcγR humanized mice (huFcγR) that express human FcγR receptors under the control of their human regulatory elements were used.37 In this model, endogenous mouse FcγR genes were deleted and human FcγRs transgenes (encoding huFcγRI, huFcγRIIAR131, huFcγRIIBI232, huFcγRIIIAF158, and huFcγRIIIB) that faithfully recapitulate the unique pattern of human FcγR expression were introduced. huFcγR mice were transplanted with EL-4 thymoma cell line expressing human CD38 transgene (supplemental Figure 3A) and were subsequently treated with injections of SAR442085, daratumumab, isatuximab, or isotype control (D1, D4, D7, D11 and D14) (Figure 3A). While isatuximab and daratumumab led to 50% survival of HuCD38-EL-4-bearing mice as compared with isotype control at the dose of 10 mg/kg, SAR442085 efficacy was higher with 90% of the mice surviving (Figure 3B). More strikingly, while isatuximab and daratumumab did not increase the survival of HuCD38-EL-4-bearing mice as compared with isotype control at a lower dose of 1.25 mg/kg, SAR442085 efficacy was maintained (Figure 3C). To better appreciate the immune changes associated with SAR442085 treatment in vivo, flow cytometry analyses were performed on splenocytes from long-term surviving mice (SAR442085, isatuximab, and daratumumab groups; 90 days) or end-point tumor-bearing isotype-treated mice. We found that SAR442085 induced an increase in the percentage and number of NK cells, macrophages, and dendritic cells compared with isotype, isatuximab, or daratumumab (Figure 3D; supplemental Figure 3B).
![SAR442085 displays a better antitumor activity than daratumumab and isatuximab in vivo. (A-D) HuCD38-EL-4 cell line was injected IV into huFcgR mice that were treated with SAR442085, daratumumab, or isatuximab (IP; 10 mg/kg [B] or 1.25 mg/kg [C]). Isotype control was used at 10 mg/kg, and the same group is represented in panels B and C. (A) Experimental design. (B-C) Kaplan-Meier survival curves showing the survival of mice after treatment with the indicated mAbs at 10 mg/kg (B) or 1.25 mg/kg (C). Statistical analyses were performed using log-rank tests and after Bonferonni Holm correction. **: P < .01 vs control group; ns = non-significant vs control; ##: P < .01 vs daratumumab; x: P < .05 vs isatuximab. (D) The percentages of the indicated splenic immune cell populations were determined for the indicated groups of mice. Data are pooled from 10 mg/kg and 1.25 mg/kg mAb treated mice. Statistical analyses were performed using Kruskal-Wallis test followed by Dunn’s tests for pairwise comparisons. P values < 0.05 were considered statistically significant. *: P < .05; ***: P < .001; ns = non-significant. (E-G) Purified NK cell from huFcgR mice were injected into Rag2−/−IL2rg−/− mice. Seven days after mice were challenged with Vk12653-huCD38 cells (2 × 106; IV) and subsequently treated as specified with 1 mg/Kg of SAR442085, daratumumab, isatuximab, or isotype. (E) Experimental design. (F-G) Representative picture (F) and Graph (G) of serum protein electrophoresis 35 days post-MM challenge. Statistical analyses were performed using Kruskal-Wallis test followed by Dunn’s tests for pairwise comparisons. *P < .05; **P < .01, ***P < .001. (H) Kaplan-Meier curves showing the evolution of paraproteinemia-free mice for the indicated groups of mice. n = 22-24 mice pooled from 5 independent experiments. Statistical analyses were performed using log-rank tests followed by a Holm-Sidak correction for multiplicity. **P < .01, ***P < .001](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/8/10.1182_blood.2021012448/5/m_bloodbld2021012448f3.png?Expires=1769089442&Signature=Gif-HmIhnzdjte7IA313PwbK2RN~XWmmMjVsKoEQt53TN9Rd155kcmePmkdCtB~3dkSRYMxwoNxuNyUZ8MYV6QYzSIaVabJ5B2mLlPUQWM9Eoi94xOT-xrtrxXBzTQdsuotwVrtKnopuxQcAqr6Oa1bK2w~0ImtnesHAXhshiaEL9v42tcDl3K6c7MRVXz8tYOnQuigr~QmwTIiYUMxO-H3yzUWct7L9yRZd15qowJFesPYxQuelUh65vpxZqOyI5L7s9wQmV5KqmAls2m13XvPBpdBw4Jf~G3tVuATx6~UXRc9Wfo~LyfDaIa6Zyv4FT29~BsbIcMAdoJhk2DC3pg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
SAR442085 displays a better antitumor activity than daratumumab and isatuximab in vivo. (A-D) HuCD38-EL-4 cell line was injected IV into huFcgR mice that were treated with SAR442085, daratumumab, or isatuximab (IP; 10 mg/kg [B] or 1.25 mg/kg [C]). Isotype control was used at 10 mg/kg, and the same group is represented in panels B and C. (A) Experimental design. (B-C) Kaplan-Meier survival curves showing the survival of mice after treatment with the indicated mAbs at 10 mg/kg (B) or 1.25 mg/kg (C). Statistical analyses were performed using log-rank tests and after Bonferonni Holm correction. **: P < .01 vs control group; ns = non-significant vs control; ##: P < .01 vs daratumumab; x: P < .05 vs isatuximab. (D) The percentages of the indicated splenic immune cell populations were determined for the indicated groups of mice. Data are pooled from 10 mg/kg and 1.25 mg/kg mAb treated mice. Statistical analyses were performed using Kruskal-Wallis test followed by Dunn’s tests for pairwise comparisons. P values < 0.05 were considered statistically significant. *: P < .05; ***: P < .001; ns = non-significant. (E-G) Purified NK cell from huFcgR mice were injected into Rag2−/−IL2rg−/− mice. Seven days after mice were challenged with Vk12653-huCD38 cells (2 × 106; IV) and subsequently treated as specified with 1 mg/Kg of SAR442085, daratumumab, isatuximab, or isotype. (E) Experimental design. (F-G) Representative picture (F) and Graph (G) of serum protein electrophoresis 35 days post-MM challenge. Statistical analyses were performed using Kruskal-Wallis test followed by Dunn’s tests for pairwise comparisons. *P < .05; **P < .01, ***P < .001. (H) Kaplan-Meier curves showing the evolution of paraproteinemia-free mice for the indicated groups of mice. n = 22-24 mice pooled from 5 independent experiments. Statistical analyses were performed using log-rank tests followed by a Holm-Sidak correction for multiplicity. **P < .01, ***P < .001
SAR442085 displays a better antitumor activity than daratumumab and isatuximab in vivo. (A-D) HuCD38-EL-4 cell line was injected IV into huFcgR mice that were treated with SAR442085, daratumumab, or isatuximab (IP; 10 mg/kg [B] or 1.25 mg/kg [C]). Isotype control was used at 10 mg/kg, and the same group is represented in panels B and C. (A) Experimental design. (B-C) Kaplan-Meier survival curves showing the survival of mice after treatment with the indicated mAbs at 10 mg/kg (B) or 1.25 mg/kg (C). Statistical analyses were performed using log-rank tests and after Bonferonni Holm correction. **: P < .01 vs control group; ns = non-significant vs control; ##: P < .01 vs daratumumab; x: P < .05 vs isatuximab. (D) The percentages of the indicated splenic immune cell populations were determined for the indicated groups of mice. Data are pooled from 10 mg/kg and 1.25 mg/kg mAb treated mice. Statistical analyses were performed using Kruskal-Wallis test followed by Dunn’s tests for pairwise comparisons. P values < 0.05 were considered statistically significant. *: P < .05; ***: P < .001; ns = non-significant. (E-G) Purified NK cell from huFcgR mice were injected into Rag2−/−IL2rg−/− mice. Seven days after mice were challenged with Vk12653-huCD38 cells (2 × 106; IV) and subsequently treated as specified with 1 mg/Kg of SAR442085, daratumumab, isatuximab, or isotype. (E) Experimental design. (F-G) Representative picture (F) and Graph (G) of serum protein electrophoresis 35 days post-MM challenge. Statistical analyses were performed using Kruskal-Wallis test followed by Dunn’s tests for pairwise comparisons. *P < .05; **P < .01, ***P < .001. (H) Kaplan-Meier curves showing the evolution of paraproteinemia-free mice for the indicated groups of mice. n = 22-24 mice pooled from 5 independent experiments. Statistical analyses were performed using log-rank tests followed by a Holm-Sidak correction for multiplicity. **P < .01, ***P < .001
To confirm these results using a preclinical mouse model mimicking myeloma pathology, VK*MYC cell line Vk1265338,39,41 was transduced with HuCD38 molecule (supplemental Figure 3C-F). This cell line generated from spontaneous MM-bearing VK*MYC mice induces the development of progressing MM lesions in the BM within 3 to 5 weeks, which are associated with progressive accumulation of serum monoclonal Ig (M spike), thus mimicking MM development in humans.38,39,41 Of note, HuCD38-Vk12653 expressed lower levels of CD38 than HuCD38-EL-4, corresponding to a CD38low tumor cell target (supplemental Figure 3A). To address specifically the role of NK cells in the higher antimyeloma potency of SAR442085, NK cells were purified from huFcγR mice and transferred into immunodeficient Rag2−/−Il2rg−/− recipient mice. These mice reconstituted with HuFcγRIIIa+ NK cells (supplemental Figure 3F) were injected 1 week later with HuCD38-Vk12653 cells and subsequently treated with isotype control, SAR442085, isatuximab, or daratumumab (1 mg/kg, D8, D10, D12, D15, D17, and D19) (Figure 3E). The 3 different anti-CD38mAbs significantly decreased serum paraproteinemia compared with isotype control (Figure 3F-G). Yet, SAR442085 had a significantly higher efficacy in limiting MM burden (Figure 3F-G) and induced better disease-free survival compared with isatuximab and daratumumab (Figure 3H). Altogether, these results confirm that SAR442085 has higher NK cell-dependent antitumor efficacy than preexisting anti-CD38 mAbs in vivo.
SAR442085 has higher ability to stimulate ADCC against primary MM cells
As MM cell lines and in vivo models do not fully reflect the tumor heterogeneity found in MM patients, we next examined the ADCC activity of SAR442085, isatuximab, and daratumumab using primary myeloma cells isolated from newly diagnosed MM patients. To this aim, NK cells isolated from HD PBMCs were incubated with freshly sorted CD138+ malignant PCs preincubated with increasing concentrations of SAR442085, isatuximab, daratumumab, or isotype control (Figure 4A). Both isatuximab, daratumumab, or SAR442085 induced a strong decrease in FcγRIIIa expression at the cell surface of NK cells in the presence of PCs incubated with high concentrations of (Figure 4B-C). These events reflecting FcγRIIIa engagement induced by the Fc portion of anti-CD38 mAbs bound to the cell surface of target cells were not observed with isotype control antibodies that do not bind CD38 (supplemental Figure 4A-B). Interestingly, in the presence of PCs incubated with low concentrations of mAbs, SAR442085 was more effective at decreasing NK cell FcγRIIIa expression than isatuximab and daratumumab (Figure 4B-C). The higher potency of SAR442085 to trigger FcγRIIIa engagement upon binding to CD38 was associated with a significant increase in NK cells expressing CD69 activation marker compared with isatuximab and daratumumab (Figure 4D-E; supplemental Figure 4C). The analysis of cytokines secreted in the coculture supernatant revealed that NK cells produced significantly higher amounts of IFN-γ and MIP1-α in the presence of MM cells coated with SAR442085 than with isatuximab and daratumumab, thus confirming the higher activating signals provided by this compound (Figure 4F). Finally, to gain insight into the killing capabilities of NK cells, we analyzed the cell surface expression of CD107a degranulation marker upon coculture with primary MM cells and the different anti-CD38 mAbs (Figure 4G-H). Our results demonstrated that the percentage of CD107a+ degranulating NK cells was significantly increased in coculture with SAR442085 coated targets as compared with isatuximab and daratumumab (Figure 4G-H). Taken together, these results indicate that SAR442085 has a higher ability than isatuximab and daratumumab to drive ADCC against primary MM cells by NK cells.
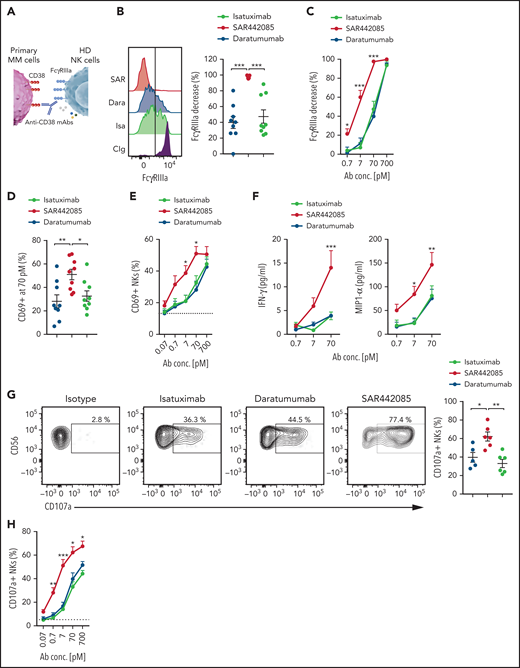
SAR442085 has higher ability to stimulate ADCC against primary MM cells. (A-H) NK cells purified from HD were cocultured with newly diagnosed MM patients' CD138+ plasma cells preincubated with increasing concentrations of SAR442085, daratumumab, or isatuximab. Isotype control was used at 700 pM in all conditions. (B) FACS histograms and graph showing the NK cell changes in FcγRIIIa expression as compared with Ig control in the presence of the indicated mAbs at 70 pM. (C) Relative decrease in FcγRIIIa expression as compared with Ig control (mean ± SEM of 9 MM patients). (D) FACS histograms and graph showing the CD69 expression of NK cells in the presence of the indicated mAbs at 70 pM. (E) CD69 expressing NK cells with increasing dose of the indicated mAbs (mean ± SEM of 9 MM patients). (F) IFN-γ and MIP1-α concentrations in the coculture supernatants after 18 hours. (G) FACS plots and graph showing the NK cell CD107a expression in the presence of the indicated mAbs at 70 pM. (H) CD107a expressing NKs with increasing dose of the indicated mAbs (mean ± SEM of 6 MM patients). Statistical differences between groups were determined by a 2-way ANOVA with Bonferroni-Holm correction. For IFN-γ and MIP1-α cytokines, this analysis was performed on squared root transformed data. *P < .05, **P < .01, ***P < .001.
SAR442085 has higher ability to stimulate ADCC against primary MM cells. (A-H) NK cells purified from HD were cocultured with newly diagnosed MM patients' CD138+ plasma cells preincubated with increasing concentrations of SAR442085, daratumumab, or isatuximab. Isotype control was used at 700 pM in all conditions. (B) FACS histograms and graph showing the NK cell changes in FcγRIIIa expression as compared with Ig control in the presence of the indicated mAbs at 70 pM. (C) Relative decrease in FcγRIIIa expression as compared with Ig control (mean ± SEM of 9 MM patients). (D) FACS histograms and graph showing the CD69 expression of NK cells in the presence of the indicated mAbs at 70 pM. (E) CD69 expressing NK cells with increasing dose of the indicated mAbs (mean ± SEM of 9 MM patients). (F) IFN-γ and MIP1-α concentrations in the coculture supernatants after 18 hours. (G) FACS plots and graph showing the NK cell CD107a expression in the presence of the indicated mAbs at 70 pM. (H) CD107a expressing NKs with increasing dose of the indicated mAbs (mean ± SEM of 6 MM patients). Statistical differences between groups were determined by a 2-way ANOVA with Bonferroni-Holm correction. For IFN-γ and MIP1-α cytokines, this analysis was performed on squared root transformed data. *P < .05, **P < .01, ***P < .001.
SAR442085 antibody trigger higher ADCC by MM patients' NK cells
Several reports suggest that NK cells from MM patients may become dysfunctional upon exposure to MM tumors.42 To address this feature, NK cells were sorted from the BM of newly diagnosed MM patients and cocultured with RPMI-8226 MM cells coated with different concentrations of anti-CD38 mAbs or isotype control (Figure 5A). FcγRIIIa engagement by MM NK cells was increased with SAR442085 for most of the tested concentrations compared with daratumumab (Figure 5B-C). The higher potency of SAR442085 to engage FcγRIIIa was confirmed by a higher percentage of degranulating CD107a+ NK cells in the presence of RPMI-8226 coated with SAR442085 compared with daratumumab, even at the highest concentrations (Figure 5D-E). Independently of the density of CD38 on target cells (high for MOLP-8 and low for KMS-12-BM), SAR442085 showed a higher potency to engage FcγRIIIa and to drive NK cell degranulation (supplemental Figure 5A-H). In addition, higher FcγRIIIa engagement was visible for NK cells from FcγRIIIa-158 V/V, F/V, and F/F MM patients demonstrating that SAR442085 increased efficacy is independent of FCGR3A polymorphism (Figure 5F).
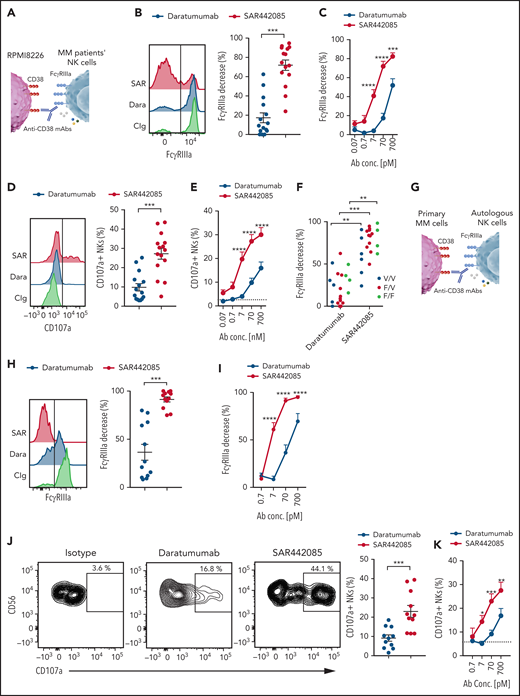
SAR442085 antibody trigger higher ADCC by MM patients' NK cells. (A-F) NK cells purified from the bone marrow of newly diagnosed MM patients were cocultured with RPMI-8226 MM cell line in the presence of daratumumab, SAR442085, or isotype control (700 pM). (A) Experimental design. (B) FACS histograms and graph showing the NK cell changes in FcγRIIIa expression as compared with Ig control in the presence of the indicated mAbs at 70 pM. (C) Relative decrease in FcγRIIIa expression as compared with Ig control induced by increasing dose of the indicated mAbs (mean ± SEM of 15 MM patients). (D) FACS histograms and graph showing the NK cell CD107a expression in the presence of the indicated mAbs at 70 pM. (E) CD107a expressing NKs with increasing dose of the indicated mAbs (mean ± SEM of 15 MM patients). (F) NK cell FcγRIIIa decrease induced by the indicated mAbs at 70 pM according to the FcγRIIIa-158 V or F genotype of the MM patients. (G-K) NK cells purified from the bone marrow of newly diagnosed MM patients were cocultured with MM patients’ CD138+ plasma cells preincubated with increasing concentrations of SAR442085, daratumumab, or isotype control (700 pM). (G) Experimental design. (H) FACS histograms and graph showing the NK cell changes in FcγRIIIa expression as compared with Ig control in the presence of the indicated mAbs at 70 pM. (I) Relative decrease in FcγRIIIa expression as compared with Ig control induced by increasing dose of the indicated mAbs (mean ± SEM of 12 MM patients). (J) FACS plots and graph showing the NK cell CD107a expression in the presence of the indicated mAbs at 70 pM. (K) CD107a expressing NKs with increasing dose of the indicated mAbs (mean ± SEM of 11 MM patients). Statistical differences between SAR442085 and daratumumab groups were determined by a two-way ANOVA with Bonferroni-Holm correction. *P < .05, **P < .01, ***P < .001.
SAR442085 antibody trigger higher ADCC by MM patients' NK cells. (A-F) NK cells purified from the bone marrow of newly diagnosed MM patients were cocultured with RPMI-8226 MM cell line in the presence of daratumumab, SAR442085, or isotype control (700 pM). (A) Experimental design. (B) FACS histograms and graph showing the NK cell changes in FcγRIIIa expression as compared with Ig control in the presence of the indicated mAbs at 70 pM. (C) Relative decrease in FcγRIIIa expression as compared with Ig control induced by increasing dose of the indicated mAbs (mean ± SEM of 15 MM patients). (D) FACS histograms and graph showing the NK cell CD107a expression in the presence of the indicated mAbs at 70 pM. (E) CD107a expressing NKs with increasing dose of the indicated mAbs (mean ± SEM of 15 MM patients). (F) NK cell FcγRIIIa decrease induced by the indicated mAbs at 70 pM according to the FcγRIIIa-158 V or F genotype of the MM patients. (G-K) NK cells purified from the bone marrow of newly diagnosed MM patients were cocultured with MM patients’ CD138+ plasma cells preincubated with increasing concentrations of SAR442085, daratumumab, or isotype control (700 pM). (G) Experimental design. (H) FACS histograms and graph showing the NK cell changes in FcγRIIIa expression as compared with Ig control in the presence of the indicated mAbs at 70 pM. (I) Relative decrease in FcγRIIIa expression as compared with Ig control induced by increasing dose of the indicated mAbs (mean ± SEM of 12 MM patients). (J) FACS plots and graph showing the NK cell CD107a expression in the presence of the indicated mAbs at 70 pM. (K) CD107a expressing NKs with increasing dose of the indicated mAbs (mean ± SEM of 11 MM patients). Statistical differences between SAR442085 and daratumumab groups were determined by a two-way ANOVA with Bonferroni-Holm correction. *P < .05, **P < .01, ***P < .001.
Finally, to evaluate the anti-MM potency of SAR442085 and daratumumab in an ex vivo translatable model, we performed experiments involving NK cells and autologous malignant plasmocytes isolated from newly diagnosed MM patients (Figure 5G). NK cell FcγRIIIa engagement and degranulation were significantly improved with SAR442085 compared with daratumumab (Figure 5H-K; supplemental Figure 5I-K). Altogether, our results demonstrate that the new anti-CD38 mAb SAR442085 enhances the ADCC activity of myeloma infiltrating NK cells against autologous targets.
Discussion
The anti-CD38 mAbs such as daratumumab, and more recently isatuximab, are among the most promising drugs recently approved for the treatment of MM. These agents showed striking activity as single agents or in combination with standard-of-care antimyeloma drugs, both for relapsed/refractory and newly diagnosed myeloma patients.43 Additionally, anti-CD38 targeting mAbs (MOR202 and TAK-079) have also been developed and are being tested in clinical trials.44,45 Still, despite the low toxicity profile and high efficacy of anti-CD38 mAb-based immunotherapy, there is a substantial heterogeneity in the quality and duration of the response among patients with all the different anti-CD38 mAbs tested so far, highlighting the need of more active anti-CD38 agents with higher response rates.43 In this study, through biochemical and functional assays, using patient samples and an in vivo model recapitulating important features of MM as progressive accumulation of serum monoclonal Ig, we demonstrated the preclinical efficacy of SAR442085, a newly developed anti-CD38 antibody with increased binding affinity to FcγRIIa and FcγRIIIa.
CDC inhibitors CD46, CD55, and CD59 are frequently overexpressed by MM primary cells, and only a small fraction of patients may be susceptible to anti-CD38 mAb-induced CDC.46 By Contrast, accumulating evidence indicates that NK cell-mediated ADCC represents a key mechanism in isatuximab and daratumumab-mediated MM cells killing in vitro and in vivo.11,47,48 Based on these observations, improving NK cell functions may represent a promising strategy to increase the efficacy of CD38-targeting mAbs. For example, IMiDs, potent activators of NK cells, were shown to potentiate ADCC mediated by CD38-directed antibodies, and recent clinical trials showed the clinical relevance of anti-CD38 mAbs and IMiDs combinations.2,49-51 Additional study evidenced that blocking NK cell inhibitory receptors, such as killer immunoglobulin receptors, significantly enhance daratumumab-mediated ADCC of primary MM cells in ex vivo assays.52 In addition, NK cell-mediated ADCC is critically dependent on the binding of the Fc region of tumor-targeting mAbs to FcγRIIIa. Increasing FcγRIIIa affinity was shown to represent a promising therapeutic strategy to improve clinical efficacy of these mAbs.26 In the present study, increased binding of SAR442085 to FcγRIIIa resulted in improved NK cell activation and killing of CD38-expressing targets compared with isatuximab and daratumumab, both in vitro and in vivo, using two different mouse models. Fc-engineered mAbs with increased affinity to the activating Fc receptor FcγRIIIa have already shown superior activity without increased toxicity in other malignancies, and many are currently approved or are in advanced clinical development.26,31-33 These evidences together with the promising preclinical results presented in the present study underline the therapeutic promise of ADCC-enhanced tumor targeting CD38 mAbs in myeloma.
The binding affinity of FcγRIIIa to IgG was shown to differ between the two main allelic variants present among the general population, with FcγRIIIa-158V having higher affinity for IgG than FcγRIIIa-158F.53 This FcγRIIIa polymorphism has been shown to impact the clinical efficacy of several tumor-targeting antibodies, such as elotuzumab,54 trastuzumab,55 or rituximab,56 and partially affects the antimyeloma efficacy of anti-CD38 mAbs.57-59 Interestingly, we found an increased affinity of SAR442085 for both FcγRIIIA F and V variants compared with daratumumab with only a slight difference in affinity for these receptor allelic variants. In addition, SAR442085 increased MM patients' NK cell activation and MM cell killing regardless of FcγRIIIa polymorphism. These results suggest that SAR442085 may have improved efficacy as compared with other anti-CD38 mAbs, especially given the high frequency of patients bearing the low affinity FcγRIIIa F/F and FcγRIIIa V/F variants. The superior efficacy of SAR442085 compared with preexisting anti-CD38 mAbs against aggressive VK*MYC and EL-4-derived tumors observed in vivo using FcγR humanized mice that express the low affinity variant FcγRIIIa F/F confirms this hypothesis.
Initial studies identified a correlation between CD38 expression on MM cells and ADCC or CDC induced by daratumumab.47 Our results indicate that increasing FcγR affinity of CD38 mAb by Fc-modification may limit the impact of CD38 density at the cell surface of target cells. Indeed, we observed a higher ability of SAR442085 to drive NK cell degranulation and target cell lysis even against MM cell lines expressing low levels of CD38, such as KMS-12-BM. In addition, despite heterogeneous expression of CD38 on primary MM cells, our results showed that SAR442085, unlike daratumumab, consistently induced high NK cell activation and degranulation against MM patient primary cells even in autologous settings or using suboptimal concentrations. These results suggest that SAR442085 may have a broader clinical efficacy than daratumumab especially in MM patients with lower CD38 expression. This hypothesis is supported by the higher NK cell-dependent antimyeloma efficacy of SAR442085 in our HuCD38-VK12653 in vivo model that expresses lower levels of CD38 than most malignant PCs from MM patients. Recent data suggest that anti-CD38 mAbs may have efficacy in other hematological malignancies, such as chronic lymphocytic leukemia, T cell acute lymphoid leukemia, and acute myeloid leukemia.60,61 However, tumor cells in these pathologies often express low levels of CD38 compared with myeloma cells, potentially limiting the therapeutic promise of these agents. Our results suggest SAR442085 may represent a good alternative against these low CD38-expressing malignancies. Importantly, SAR442085 may have therapeutic utility in MM patients who have relapsed or are refractory to daratumumab and isatuximab, as these treatments induce a decrease in CD38 levels.62 Although our study focused mainly on the FcγRIIIa-mediated ADCC by NK cells, the phagocytosis of antibody-opsonized tumor cells may also play an important role in the efficacy of SAR442085, as it is the case for most anti-CD38 mAbs.12,13 Specifically, the higher SAR442085 affinity to FcγRIIa and the higher ADCP observed in our assays suggest that SAR442085 could have an increased ADCP-mediated killing potency compared with other existing tumor targeting antibodies. Once again, this feature of SAR442085 suggests broader spectrum efficacy of this agent compared with preexisting anti-CD38 mAbs.
Comparing the ADCC efficacy of different mAbs in vivo is often complicated by the differences existing between murine and human Fc receptors. In mice, antibody-dependent cytotoxicity is primarily mediated by both FcγRIII and FcγRIV, while in humans it is predominantly exerted through FcγRIIIa expressed on NK cells. Moreover, the antibody isotypes that better induce cytotoxicity in mice and humans are different. While murine IgG2a is the best cytotoxicity inducer-antibody isotype, IgG1 is usually the best human ADCC inducer. To bypass these issues, we used FcγR humanized mice (huFcγR) that express human FcγR under the control of their human regulatory elements. We first used EL-4 expressing human CD38 and found that SAR442085 significantly increased the survival of CD38-EL4-bearing huFcγR mice compared with isatuximab and daratumumab. The higher NK cell-dependent anti-MM potency of SAR442085 was then confirmed using huFcγR NK cell adoptive transfer and the most relevant myeloma mouse model, VK*MYC overexpressing human CD38. Taken together, these results highlight the higher potency of SAR442085 to drive NK cell-dependent tumor myeloma killing in vivo. Still, additional immune mechanisms may account for the efficacy of anti-CD38 mAbs in myeloma patients. For example, the oligoclonal expansion of CD8+ cytotoxic T cells seems to represent an important determinant of the therapeutic efficacy of anti-CD38 mAbs.17-19 In addition, CD38 is expressed at high levels on several immunosuppressive cells, such as myeloid-derived suppressor cells and regulatory T and B cells (Tregs and Bregs), and treatment with daratumumab was shown to decrease these suppressor cells in the BM microenvironment, resulting in an increased T-cell clonality. Similarly, isatuximab was shown to limit Treg numbers and to decrease their immunosuppressive functions.18 Whether these immunomodulatory effects are also enhanced by SAR442085 remains an open question.
Taken together, these results establish that SAR442085 has higher antitumor efficacy than preexisting anti-CD38 mAb and support the current evaluation of this next-generation anti-CD38 antibody in phase 1 clinical trial for patients with relapsed/refractory MM (NCT04000282). While this trial will primarily focus on the safety and determination of recommended dose, preliminary efficacy results in both anti-CD38 mAb pretreated (ie, monotherapy tumor response in daratumumab failure) and anti-CD38 mAb naïve MM (ie, monotherapy tumor response rate above historically observed response rate with daratumumab) will be of interest to guide the next clinical development steps. Observation of antitumor activity in MM may allow to expand the clinical investigation to other hematologic malignancies with lower CD38 expression.
Acknowledgments
The authors are grateful to their health care professionals during the COVID-19 crisis. They thank Manon Farcé and members from the core facilities at the Cancer Research Center of Toulouse for their help. L.M. was supported by “la Fondation ARC” (PGA1-20160203788 and 20190208630), by the “Institut National du Cancer” (INCA, PLBIO R16100BB, R19-045 and R20-229), Cancer Research Institute/Bristol-Myers Squibb CLIP Grant, and the Fondation Toulouse cancer santé and IUCT-O translational research program. The authors thank Jeffrey V. Ravetch for giving them access to the humanized FcgR mice model. The authors would also like to thank Marie Gagnaire for performing Caliper binding assay studies, Laurent Bassinet and Virginie Boisrobert-Dheilly for performing cellular studies, Laure-Marie Meyer et Ravi Rangara for animal pharmacological studies, Fanny Windenberger for the statistical analyses, and all members of Sanofi Immuno-Oncology Research, Pharmacology, and Biologics Research teams who contributed to this project.
Authorship
Contribution: Study conception and design: S.K., B.K.D., N.E.-M., A.T., A.F., N.C, C.N., J.-L.T., M. Chiron, L.M., and A.V.-O.; Acquisition of data: S.K., B.K.D., N.E.-M., H.B., A.T., N.C, I.A., C.N., and M. Cuisinier; Analysis and interpretation of data: S.K., B.K.D., N.E.-M., A.T., H.B., C.N., A.F., J.-L.T., M. Chiron, L.M., and A.V.-O.; Drafting of manuscript: N.E-M., C.N., M. Chiron, L.M., and A.V.-O.; Critical revision and editing: N.E-M., A.T., A.F., H.B., C.N., S.S.S., H.v.d.V., D.W., M. Chiron, L.M., and A.V.-O.; and Provision of key materials: A.F., C.N., J.C, S.S.S., A.V.-O., and H.A.-L.
Conflict-of-interest disclosure: L.M., H.A.-L and S.K. had a research agreement with Sanofi-Aventis between 2016 and 2020. J.-L.T. received a grant from Sanofi-Aventis during the conduct of the study and reports being a former consultant for Sanofi. B.K.D was a Sanofi employee funded by a CIFRE doctoral fellowship (N°2015/0834) from ANRT. N.E.-M., A.T., A.F., H.B., C.N., I.A., S.S.S., H.v.d.V., D.W., M. Cuisinier, and A.V.-O. are Sanofi employees and shareholders.
Correspondence: Ludovic Martinet, INSERM UMR 1037, Cancer Research Center of Toulouse, 2 av Hubert Curien, 31037 Toulouse, France; e-mail: ludovic.martinet@inserm.fr; and Angela Virone-Oddos, Sanofi Oncology Research, 13 quai Jules Guesde, 94403 Vitry-sur-Seine, France; e-mail: Angela.Virone-Oddos@sanofi.com
Send data sharing requests via e-mail to the corresponding authors.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal