

Key Points
The CLL2-GIVe trial tested the triple combination with ibrutinib, venetoclax, and obinutuzumab in CLL with del(17p) and/or TP53 mutation
The CLL2-GIVe regimen has a manageable safety profile and shows promise as an effective, fixed duration, first-line treatment of high-risk CLL

Abstract
Despite considerable treatment advances with targeted therapies for patients with chronic lymphocytic leukemia (CLL) deemed high-risk [del(17p) and/or TP53 mutation], the outcome is still inferior compared with other CLL patients. Combining multiple agents with distinct mechanisms of action may further improve outcomes. CLL2-GIVe is an open-label, multicenter trial which enrolled patients with previously untreated CLL with del(17p) and/or TP53 mutation. Patients received induction therapy with obinutuzumab (GA-101), ibrutinib, and venetoclax (GIVe) for cycles 1 through 6 and consolidation therapy with venetoclax and ibrutinib for cycles 7 through 12. Ibrutinib monotherapy was continued for cycles 13 through 36 in patients not reaching a complete response (CR) with serial undetectable minimal residual disease (uMRD) after consolidation. The primary endpoint was CR rate at cycle 15 (final restaging). Secondary endpoints included MRD, survival, and safety. All 41 patients enrolled between September 2016 and August 2018 received study treatment and were included in efficacy and safety populations. With a CR rate of 58.5% at cycle 15, the primary endpoint was met (95% CI: 42.1-73.7; P < .001). At final restaging, 78.0% of patients had uMRD in peripheral blood (PB); 65.9% of patients had uMRD in bone marrow (BM). Estimated progression-free survival (PFS) and overall survival (OS) rates at 24 months were both 95.1%. Adverse events were reported in all patients; most were low grade (grade ≥3: 23.9%). Two deaths were reported (cardiac failure and ovarian carcinoma), neither related to study treatment. The CLL2-GIVe treatment regimen has a manageable safety profile and is a first-line treatment of good efficacy for patients with high-risk CLL.
Introduction
Chronic lymphocytic leukemia (CLL) is the most common leukemia in the Western world and affects mainly elderly patients.1 Chemoimmunotherapy has been the standard of care, but its limitations include adverse events (AEs) and reduced efficacy, in particular in high-risk subgroups.2-5
The use of novel, targeted agents resulted in marked improvement in prognosis for most CLL patient subgroups.6-12 In particular, data from several phase 3 trials, which mainly investigated novel agents (Bruton tyrosine kinase inhibitors [BTKi] ibrutinib, acalabrutinib, BCL-2 inhibitor venetoclax, and CD20 monoclonal antibody obinutuzumab) have consistently confirmed their efficacy, showing improvements in progression-free survival (PFS) in direct comparison with chemoimmunotherapy regimens13-20 with overall survival (OS) increase in some studies.15,16 Despite this, the prognosis for patients with high-risk CLL [del(17p) and/orTP53 mutation] is still inferior, with lower remission rates, shorter remission duration, and inferior survival compared with patients without these genetic risk factors.21-34 Ibrutinib and venetoclax are approved for first-line treatment of CLL in combination with obinutuzumab.35,36 The ESMO (European Society for Medical Oncology) guidelines recommend venetoclax monotherapy, obinutuzumab/venetoclax or ibrutinib for the treatment of patients with del(17p) and/or TP53 mutation.37
The CLL2-GIVe trial combines obinutuzumab (GA-101), ibrutinib, and venetoclax (GIVe) with distinct mechanisms of action and investigates the safety and efficacy of this time-limited combination therapy in patients with high-risk CLL, with the aim of achieving more durable and deeper remissions.
Patients and methods
CLL2-GIVe (NCT02758665) is an ongoing open-label, multicenter trial conducted at 11 German centers. The trial was approved by the institutional review board at each participating institution and was conducted in accordance with the Declaration of Helsinki and International Conference on Harmonization Guidelines for Good Clinical Practice. All patients provided written informed consent. Here we report the primary endpoint analysis with a data cutoff date of 17 July 2020.
Eligibility
Eligible patients were aged ≥18 years with previously untreated CLL and del(17p) and/or TP53 mutation requiring treatment, diagnosed according to the International Workshop on CLL (iwCLL) criteria.38,39 Diagnosis and need for treatment were determined by the treating clinician and confirmed during the central screening according to iwCLL criteria.38,40 During central review, patients were defined as fit (adequate organ function, normal creatinine clearance rate, and a Cumulative Illness Rating Scale [CIRS] score ≤6) or unfit (creatinine clearance <50 mL/min and/or CIRS >6).38,40 Patients with transformation of CLL (ie, Richter transformation, prolymphocytic leukemia), known central nervous system involvement, and/or a history of progressive multifocal leukoencephalopathy were excluded from the study. Additional eligibility criteria are summarized in the supplemental Appendix.
Study design and treatments
The treatment consists of an induction phase with the triple combination of obinutuzumab, venetoclax, and ibrutinib, a consolidation phase with ibrutinib and venetoclax (6 cycles each), and a maintenance phase with ibrutinib monotherapy according to response and minimal residual disease (MRD) level (Figure 1). During induction, obinutuzumab was administered IV for 6 cycles (1 cycle = 28 days), starting with 100 mg on day 1 and 900 mg on day 2 (or 1000 mg on day 1), 1000 mg on day 8, and 1000 mg on day 15 of cycle 1, and subsequently 1000 mg on day 1 of cycles 2 through 6. Ibrutinib 420 mg daily was administered orally from day 1 of cycles 1 through 15. The daily oral venetoclax regimen was initiated on day 22 of cycle 1, starting with a 5-week dose ramp-up (1 week each of 20, 50, 100, and 200 mg, then 400 mg daily for 1 week), thereafter continuing at 400 mg daily until completion of cycle 12.
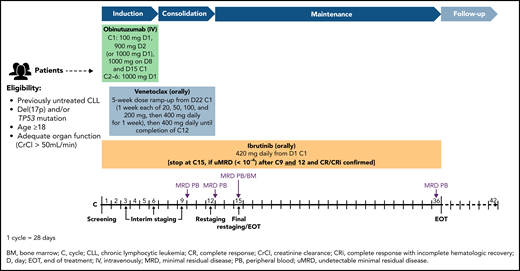
Study design and treatment schedule. The treatment is separated into an induction phase with the triple combination of obinutuzumab, venetoclax, and ibrutinib; a consolidation phase with ibrutinib/venetoclax, 6 cycles each; and a maintenance phase with ibrutinib monotherapy according to response (MRD at cycle 9, 12, 15; CT scan at cycle 12; BM biopsy during cycle 14/at cycle 15). In case of MRD negativity at cycles 9 and 12 and confirmed CR/CRi at final restaging, ibrutinib is stopped.
Study design and treatment schedule. The treatment is separated into an induction phase with the triple combination of obinutuzumab, venetoclax, and ibrutinib; a consolidation phase with ibrutinib/venetoclax, 6 cycles each; and a maintenance phase with ibrutinib monotherapy according to response (MRD at cycle 9, 12, 15; CT scan at cycle 12; BM biopsy during cycle 14/at cycle 15). In case of MRD negativity at cycles 9 and 12 and confirmed CR/CRi at final restaging, ibrutinib is stopped.
MRD in peripheral blood (PB) was measured centrally during interim staging after cycles 9 and 12, as well as at the final restaging (cycle 15). If at final restaging patients had reached a complete remission with or without complete hematologic recovery (CR/CRi) according to iwCLL criteria and had undetectable PB MRD (uMRD) in cycles 9 and 12, ibrutinib maintenance therapy was stopped. Otherwise, ibrutinib was continued until cycle 36. Beyond cycle 36, ibrutinib could be indefinitely continued (off-study) at the investigator’s discretion.
Endpoints and assessments
The primary endpoint was the CR rate at cycle 15, defined as the proportion of patients with a CR/CRi as best response according to iwCLL 2008 criteria from treatment initiation to cycle 15 (day 1).38 Secondary endpoints included the proportion of patients free from progressive disease (PD) after 12 cycles of therapy, overall response rate (ORR), ORR at end of maintenance, MRD in PB at cycle 9, 12, and 15, and bone marrow (BM) at cycle 15, time-to-event endpoints (PFS, OS, event-free survival, duration of response, treatment-free survival, and time to next CLL treatment), evaluation of subsequent treatment of CLL, and safety parameters.
Baseline assessments conducted at screening included immunophenotyping of circulating lymphocytes, central analysis of genomic aberrations with fluorescence in situ hybridization, mutational analysis of immunoglobulin heavy chain variable (IGHV) and TP53 by Sanger sequencing, and evaluation of lymph node size by physical assessment and computed tomography scanning or magnetic resonance imaging. The risk of tumor lysis syndrome (TLS) was assessed on the basis of absolute lymphocyte count (ALC) and lymph node size; patients with increased TLS risk received prophylaxis, and hospitalization was recommended for patients with a large tumor burden (defined by lymph nodes >10 cm or ALC >50 × 109/L) or those patients deemed as high risk by the treating physician.
Response was assessed at cycles 4, 6, 9, 12, and 15 (final restaging) and every 3 cycles thereafter, up to 6 months after cycle 36. MRD was analyzed centrally in accordance with international guidelines.41,42 PB MRD monitoring was performed at baseline and cycles 9, 12, 15, and 36. In patients with a treatment response, MRD in BM was assessed during cycle 14 before the final restaging at cycle 15, day 1. MRD was quantified by 4-color flow cytometry as previously validated against the use of allele-specific oligonucleotide polymerase chain reaction. MRD values were categorized into 3 levels: undetectable MRD (<10−4; ie, <1 CLL cell per 10 000 leukocytes), intermediate MRD-positive (MRD+) (≥10−4 and <10−2), and high-MRD+ (≥10−2). The median observation time is 26.6 months (range, 3.7 to 41.6).
Safety was monitored throughout treatment and every 3 to 6 months thereafter to a maximum of 42 months by monitoring and recording all AEs and serious AEs (SAEs), including laboratory evaluations and physical examination. AEs were graded using National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0 and coding of AEs was performed using the Medical Dictionary for Regulatory Activities (MedDRA) classification system. Dose reductions of ibrutinib due to grade 4 hematologic disorders were mandated by protocol; medication was stopped, and supportive measures (eg, granulocyte-colony stimulating factor [G-CSF], transfusions) were given until improvement of the disorder(s) to grade 1 to 3. Venetoclax was continued in patients with grade 4 neutropenia if recovery to grade 1 to 3 was reached within 1 week by supportive measures such as G-CSF. Otherwise, venetoclax dose reductions were required. In patients with grade 4 anemia or thrombocytopenia, venetoclax was reduced by 1 dose level (400 mg to 200 mg, 200 mg to 100 mg, 100 mg to 50 mg per day). After improvement to grade 1 to 3, the dose was increased weekly (supplemental Figure 1).
Statistical analysis
The sample size was determined to investigate the efficacy of the GIVe regimen with regard to the primary endpoint of CR rate at cycle 15. The CR rate was tested against the null hypothesis of 25% using a 2-sided one-sample binomial test. The GIVe regimen was expected to lead to an improvement of the CR rate to at least 50%. Enrollment of 36 patients was required to have a power of 90% at a 2-sided significance level of 5%; thus, by including an expected drop-out rate of 10%, we aimed to enroll 40 analyzable patients. Sample size calculations were performed with EAST 6 software and validated with nQuery Advisor.
Efficacy analyses (using IBM SPSS Statistics 27) were conducted with regard to the primary endpoint and secondary endpoints (response to therapy, MRD, PFS, OS, and evaluation of subsequent treatments) using the efficacy population, which included all enrolled patients who received at least 2 complete treatment cycles according to the intention to treat principle. Frequencies and corresponding percentages, including 95% Clopper-Pearson confidence intervals (95% CI), were reported for the primary endpoint. Kaplan-Meier estimates were reported for time-to-event secondary efficacy endpoints. Additionally, exploratory subgroup analysis of PFS according to TP53 mutational status was performed. Analyses of other secondary endpoints are planned for later data cuts when more mature data are available.
Safety analyses (using IBM SPSS Statistics 27) were conducted using the safety population, which comprised all enrolled patients who received at least 1 dose of the study drug. Relative frequencies of complete-case and per-patient AEs were reported for all categorical variables.
Results
Patients
A total of 41 patients with high-risk CLL were enrolled between September 2016 and August 2018, all of whom received the study treatment and were included in the efficacy and safety populations. One patient discontinued treatment during the induction (GIVe) phase due to a fatal AE (ovarian carcinoma, retrospectively preexisting at enrollment). Overall, 40 patients (97.6%) started consolidation therapy (venetoclax-ibrutinib) and comprised the analysis population for safety during the consolidation phase. Six patients (14.6%) discontinued treatment during the consolidation phase (1 death due to cardiac failure [preexisting mitral valve insufficiency], 3 due to AEs, 2 due to MRD negativity) (Figure 2); 34 patients (82.9%) started maintenance therapy and comprised the analysis population for safety during the maintenance phase. Twelve patients (29.3%) discontinued during cycle 14 (maintenance phase; according to protocol, final restaging examination [BM biopsy and CT scan] were done during cycle 14): 1 due to the patient’s refusal of treatment/failure to cooperate, 11 due to MRD negativity, among them 9 with CR/CRi, thus 22 patients (53.7%) completed at least 15 cycles of therapy. Sixteen patients (39.0%) discontinued treatment following cycle 15, day 1 (12 due to MRD negativity and CR, 1 due to MRD negativity and CRi, 1 due to AE, 2 due to MRD negativity and partial remission [PR] [supplemental Table 5B]), 1 patient (2.4%) discontinued at the end of treatment (end of maintenance, cycle 35) and 5 patients (12.2%) are still on treatment (Figures 2 and 3).
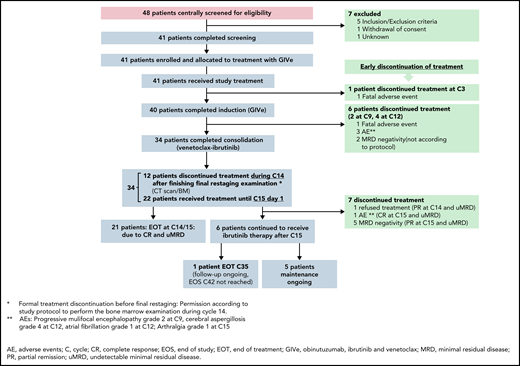
Screening, treatment, and follow-up. Of 41 screened and treated patients, 34 completed the induction and consolidation phase. Seven patients discontinued treatment according to described reasons. Of the 34 patients, final restaging was performed during cycle 14 until cycle 15, day 1. Twenty-two patients reached CR at final restaging and stopped treatment (1 patient stopped due to AE, arthralgia I°, but reaching CR at final restaging). Six patients continued ibrutinib in maintenance, 6 patients discontinued due to different described reasons. At time of data cut-off, 1 patient reached EOT.
Screening, treatment, and follow-up. Of 41 screened and treated patients, 34 completed the induction and consolidation phase. Seven patients discontinued treatment according to described reasons. Of the 34 patients, final restaging was performed during cycle 14 until cycle 15, day 1. Twenty-two patients reached CR at final restaging and stopped treatment (1 patient stopped due to AE, arthralgia I°, but reaching CR at final restaging). Six patients continued ibrutinib in maintenance, 6 patients discontinued due to different described reasons. At time of data cut-off, 1 patient reached EOT.
![Treatment, treatment discontinuation, follow-up, and genetics. Swimmer’s plot of patients separated into 3 groups [del(17p) and TP53 mutated, unmutated TP53 and del(17p), and sole TP53 mutation]. Additionally, in green dots at the left, complex karyotypes (n = 4); in blue dots, highly complex karyotypes (n = 20) are marked. Red lines indicate patients under treatment, blue lines show patients in follow-up. Treatment discontinuation is indicated in 3 ways: black asterisks as regular discontinuation, red dots as irregular discontinuation, and 2 black lines as early discontinuation. Two deaths are indicated by crosses. Six progressions are indicated in red during follow-up (between cycles 24 and 34).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/9/10.1182_blood.2021013208/4/m_bloodbld2021013208f3.png?Expires=1769078747&Signature=ClER967g3iTCLvvtxMtgCsDd3YiqbXyUdrGxS-jseBYVKgZU9MpU-elZYVeup6rDPsfIXOgRqkXUzlyNAUXlJ56NvnNBnTYzigr6FOjn1ngyzo5IHDABpoStDideFzv9t7NJQbQTgDnazcR8E7WfvA9MpN3cJElYTManVsp9ttKXyBi-RJHtoBLjwsVQNGuAiCE6p9KUDoDtY1S5H4O3~kddbnrf2nGKSlijAyk5Mph4aiGax4zXiJ2sIsPuIJgI-faJ2QY~JieXRYDfdj63nhqxfGaWi5dyzmiVFMXVinRbh16DebKQcFQds6q7x9cafYf90iJ1z6ldmHDmgcgWEA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Treatment, treatment discontinuation, follow-up, and genetics. Swimmer’s plot of patients separated into 3 groups [del(17p) and TP53 mutated, unmutated TP53 and del(17p), and sole TP53 mutation]. Additionally, in green dots at the left, complex karyotypes (n = 4); in blue dots, highly complex karyotypes (n = 20) are marked. Red lines indicate patients under treatment, blue lines show patients in follow-up. Treatment discontinuation is indicated in 3 ways: black asterisks as regular discontinuation, red dots as irregular discontinuation, and 2 black lines as early discontinuation. Two deaths are indicated by crosses. Six progressions are indicated in red during follow-up (between cycles 24 and 34).
Treatment, treatment discontinuation, follow-up, and genetics. Swimmer’s plot of patients separated into 3 groups [del(17p) and TP53 mutated, unmutated TP53 and del(17p), and sole TP53 mutation]. Additionally, in green dots at the left, complex karyotypes (n = 4); in blue dots, highly complex karyotypes (n = 20) are marked. Red lines indicate patients under treatment, blue lines show patients in follow-up. Treatment discontinuation is indicated in 3 ways: black asterisks as regular discontinuation, red dots as irregular discontinuation, and 2 black lines as early discontinuation. Two deaths are indicated by crosses. Six progressions are indicated in red during follow-up (between cycles 24 and 34).
Patient demographics and baseline characteristics are listed in Table 1. Median age was 62 years (range, 35 to 85), and median CIRS was 3 (range, 0 to 8). Overall, 32 patients (78.0%) had unmutated IGHV, 24 patients (58.5%) had both del(17p) and TP53 mutation, 15 patients (36.6%) had only TP53 mutation, and 2 patients (4.9%) had only del(17p). TP53 variants and variant allele frequencies (cutoff >10%43) and del(17p) with a proportion of affected cells are shown in the supplementary appendix (supplemental Table 2). Mutated NOTCH1 was seen in 3 patients (7.3%), while 7 patients (17.1%) had mutated SF3B1. Of the 39 patients with available information on karyotype, 15 (38.5%) did not have complex karyotype (<3 chromosomal abnormalities), 4 (10.3%) had complex karyotype (≥3 chromosomal abnormalities), and 20 (51.3%) had highly complex karyotype (≥5 chromosomal abnormalities). Prior to the initiation of treatment, 39 of the 41 patients (95.1%) had an increased risk of TLS (lymph nodes >10 cm or ALC >50 × 109/L).
Patient demographic and disease characteristics at baseline
| Characteristic | Patients (n = 41) |
|---|---|
| Median age, years (range) | 62.0 (35-85) |
| Age >65 y, n (%) | 14 (31.4) |
| Sex, n (%) | |
| Female | 17 (41.5) |
| Male | 24 (58.5) |
| Median time since first diagnosis, months (range) | 20.1 (1.1-131.6) |
| Binet stage, n (%) | |
| A | 9 (22.0) |
| B | 17 (41.5) |
| C | 15 (36.6) |
| Rai stage, n (%) | |
| 0 | 3 (7.3) |
| I | 14 (34.1) |
| II | 7 (17.1) |
| III | 11 (26.8) |
| IV | 6 (14.6) |
| B-symptoms, n (%) | |
| No | 26 (63.4) |
| Yes | 15 (36.6) |
| Median CIRS score (range) | 3 (0-8) |
| CIRS group, n (%) | |
| ≤6 | 39 (95.1) |
| >6 | 2 (4.9) |
| Median creatinine clearance, mL/min (range) | 81.1 (36.9-214.2) |
| Creatinine clearance <70 mL/min, n (%) | 15 (36.6) |
| del(17p)* and TP53 status, n (%) | |
| del(17p), TP53 mutated | 24 (58.5) |
| del(17p), TP53 unmutated | 2 (4.9) |
| No del(17p), TP53 mutated | 15 (36.6) |
| del(11q)*, n (%) | 5 (12.2) |
| del(13q)*, n (%) | 7 (17.1) |
| Trisomy 12*, n (%) | 2 (4.9) |
| IGHV mutational status, n (%) | |
| Unmutated | 32 (78.0) |
| Mutated | 6 (14.6) |
| Nonevaluable | 3 (7.3) |
| NOTCH1 status, n (%) | |
| Unmutated | 38 (92.7) |
| Mutated | 3 (7.3) |
| SF3B1 status, n (%) | |
| Unmutated | 34 (82.9) |
| Mutated | 7 (17.1) |
| Median IgG, g/L (range) | 7.5 (2.4-19.6) |
| Median IgA, g/L (range) | 0.8 (0.1-5.0) |
| Median IgM, g/L (range) | 0.4 (0.1-22.5) |
| Median IgE, g/L (range)† | 3.5 (0.3-401.1) |
| ECOG PS, n (%) | |
| 0 | 28 (68.3) |
| 1-2 | 13 (31.7) |
| TLS risk category at screening, n (%) | |
| Increased | 39 (95.1) |
| Not increased | 2 (4.9) |
| ALC at screening, n | 40 |
| Median | 65.7 |
| Range | 3.8-346.2 |
| TLS high risk: ALC >50 G/l at screening (according to protocol), n (%) | 23 (57.5) |
| TLS high risk: ALC >25 G/l at screening, n (%) | 31 (77.5) |
| Laboratory TLS (according to Cairo Bishop criteria), n | 4 |
| Urin acid (≥476 µmol/l or 25% increase from baseline), n | 2 |
| Potassium (≥6.0 mmol/l or 25% increase from baseline), n | 2 |
| Phosphorus (≥1.45 mmol/l or 25% increase from baseline), n | 4 |
| Calcium (<1.75 mmol/l or 25% increase from baseline),n | 0 |
| LDH (increased ≥25%), n | 1 |
| Serum Creatinine (increased ≥25%), n | 1 |
| Number of patients with hospitalization during dose ramp up of venetoclax, n (%) | 37 (90.2) |
| CLL-IPI risk group, n (%) | 39 |
| Low | 0 (0.0) |
| Intermediate | 0 (0.0) |
| High | 4 (10.3) |
| Very High | 35 (89.7) |
| Complex karyotype, n (%) | 39 |
| Noncomplex karyotype (<3 aberrations) | 15 (38.5) |
| Complex karyotype (≥3 aberrations) | 4 (10.3) |
| Highly complex karyotype (≥5 aberrations) | 20 (51.3) |
| Median serum β2-microglobulin, mg/dL (range)†† | 4.4 (2.5-12.5) |
| Median serum thymidine kinase, U/L (range)†† | 48.5 (9.6-300.0) |
| Characteristic | Patients (n = 41) |
|---|---|
| Median age, years (range) | 62.0 (35-85) |
| Age >65 y, n (%) | 14 (31.4) |
| Sex, n (%) | |
| Female | 17 (41.5) |
| Male | 24 (58.5) |
| Median time since first diagnosis, months (range) | 20.1 (1.1-131.6) |
| Binet stage, n (%) | |
| A | 9 (22.0) |
| B | 17 (41.5) |
| C | 15 (36.6) |
| Rai stage, n (%) | |
| 0 | 3 (7.3) |
| I | 14 (34.1) |
| II | 7 (17.1) |
| III | 11 (26.8) |
| IV | 6 (14.6) |
| B-symptoms, n (%) | |
| No | 26 (63.4) |
| Yes | 15 (36.6) |
| Median CIRS score (range) | 3 (0-8) |
| CIRS group, n (%) | |
| ≤6 | 39 (95.1) |
| >6 | 2 (4.9) |
| Median creatinine clearance, mL/min (range) | 81.1 (36.9-214.2) |
| Creatinine clearance <70 mL/min, n (%) | 15 (36.6) |
| del(17p)* and TP53 status, n (%) | |
| del(17p), TP53 mutated | 24 (58.5) |
| del(17p), TP53 unmutated | 2 (4.9) |
| No del(17p), TP53 mutated | 15 (36.6) |
| del(11q)*, n (%) | 5 (12.2) |
| del(13q)*, n (%) | 7 (17.1) |
| Trisomy 12*, n (%) | 2 (4.9) |
| IGHV mutational status, n (%) | |
| Unmutated | 32 (78.0) |
| Mutated | 6 (14.6) |
| Nonevaluable | 3 (7.3) |
| NOTCH1 status, n (%) | |
| Unmutated | 38 (92.7) |
| Mutated | 3 (7.3) |
| SF3B1 status, n (%) | |
| Unmutated | 34 (82.9) |
| Mutated | 7 (17.1) |
| Median IgG, g/L (range) | 7.5 (2.4-19.6) |
| Median IgA, g/L (range) | 0.8 (0.1-5.0) |
| Median IgM, g/L (range) | 0.4 (0.1-22.5) |
| Median IgE, g/L (range)† | 3.5 (0.3-401.1) |
| ECOG PS, n (%) | |
| 0 | 28 (68.3) |
| 1-2 | 13 (31.7) |
| TLS risk category at screening, n (%) | |
| Increased | 39 (95.1) |
| Not increased | 2 (4.9) |
| ALC at screening, n | 40 |
| Median | 65.7 |
| Range | 3.8-346.2 |
| TLS high risk: ALC >50 G/l at screening (according to protocol), n (%) | 23 (57.5) |
| TLS high risk: ALC >25 G/l at screening, n (%) | 31 (77.5) |
| Laboratory TLS (according to Cairo Bishop criteria), n | 4 |
| Urin acid (≥476 µmol/l or 25% increase from baseline), n | 2 |
| Potassium (≥6.0 mmol/l or 25% increase from baseline), n | 2 |
| Phosphorus (≥1.45 mmol/l or 25% increase from baseline), n | 4 |
| Calcium (<1.75 mmol/l or 25% increase from baseline),n | 0 |
| LDH (increased ≥25%), n | 1 |
| Serum Creatinine (increased ≥25%), n | 1 |
| Number of patients with hospitalization during dose ramp up of venetoclax, n (%) | 37 (90.2) |
| CLL-IPI risk group, n (%) | 39 |
| Low | 0 (0.0) |
| Intermediate | 0 (0.0) |
| High | 4 (10.3) |
| Very High | 35 (89.7) |
| Complex karyotype, n (%) | 39 |
| Noncomplex karyotype (<3 aberrations) | 15 (38.5) |
| Complex karyotype (≥3 aberrations) | 4 (10.3) |
| Highly complex karyotype (≥5 aberrations) | 20 (51.3) |
| Median serum β2-microglobulin, mg/dL (range)†† | 4.4 (2.5-12.5) |
| Median serum thymidine kinase, U/L (range)†† | 48.5 (9.6-300.0) |
CLL-IPI, International Prognostic Index for Chronic Lymphocytic Leukemia; ECOG PS, Eastern Cooperative Oncology Group performance status; Ig, immunoglobin.
39 of 41 patients included in these analyses.
Analyzed for 35/41 patients.
Based on hierarchical model according to Sharman et al.21
Efficacy
Overall, 24 of the 41 patients (58.5%; 95% CI, 42.1-73.7; P < .001) had a CR/CRi at final restaging at day 1 of cycle 15 (17 [41.5%] CR and 7 [17.1%] CRi); 14 patients (34.1%) achieved a PR. Three patients (7.3%) were not assessable for response at final restaging because they stopped treatment prior to cycle 12 (2 fatal AEs, 1 due to AE). For these patients, the last response assessment at interim staging (PR for all 3 patients) was considered the final restaging response; therefore, the ORR was 100% (24 CR/CRi and 17 PR) (Figure 4). With 24 complete responders and a 58.5% CR/CRi rate, the null hypothesis was rejected.
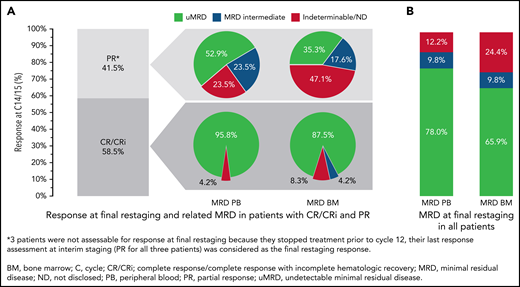
Response and MRD at final restaging (C14/15). (A) Clinical and MRD response at final restaging in patients with CR/CRi and PR. (B) MRD in PB and BM at final restaging in all patients. Percentages of patients reaching CR and PR at final restaging (cycle 14/15) and MRD in PB and BM sorted by remission status (A) and MRD response in PB and BM in all patients (B).
Response and MRD at final restaging (C14/15). (A) Clinical and MRD response at final restaging in patients with CR/CRi and PR. (B) MRD in PB and BM at final restaging in all patients. Percentages of patients reaching CR and PR at final restaging (cycle 14/15) and MRD in PB and BM sorted by remission status (A) and MRD response in PB and BM in all patients (B).
After cycles 9 and 12, 87.8% and 85.4% of all patients, respectively, had uMRD. At final restaging (cycle 15), 32 of 41 patients (78.0%) had uMRD in PB, while 27 of 41 patients (65.9%) had uMRD in BM. Most patients with CR at final restaging also had uMRD in PB (23 of 24 patients, 95.8%) and in BM (21 of 24 patients, 87.5%) (Figure 4). In patients with PR, 9 of 17 (52.9%) had uMRD in PB, and 6 of 17 (35.3%) had uMRD in BM (Figure 4). All patients with detectable MRD in PB and BM were intermediate-MRD+; no patient had high-MRD+. Three patients had MRD conversion from uMRD to intermediate MRD while on treatment: 1 patient with uMRD at cycle 9 changed to intermediate MRD at cycles 12 and 15, and 2 patients with uMRD at cycles 9 and 12 changed to intermediate MRD at cycle 15.
After a median observation time of 26.6 months (range, 3.7 to 41.6), there were 8 events (19.5%) for PFS and 2 events (4.9%) for OS. PFS and OS rates at 24 months were 95.1% (Figure 5). Median PFS was 33.5 months, and median OS was not reached. Exploratory analysis indicated that patients with both del(17p) and TP53 mutation (n = 24) had shorter PFS with 7 events and a 24-month PFS rate of 91.7% as compared with patients who had TP53 mutation but no del(17p) (n = 15, zero events; 24-month PFS rate: 100%) (supplemental Figure S2). Patient numbers were small, and events were limited; thus, regression analysis could not be performed. Other secondary endpoints are shown in supplemental Figure 5A-C and supplemental Table 4.
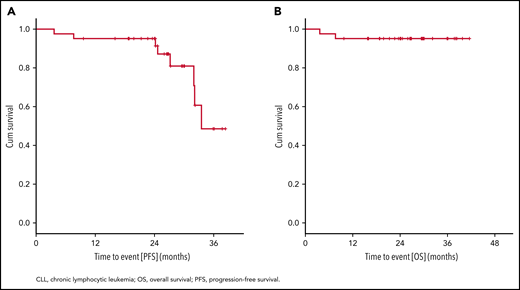
Kaplan-Meier estimates of PFS and OS. PFS (A) with 8 events after a median observation time of 26.6 months (range, 3.7 to 41.6) and OS (B) with 2 events. Median PFS is 33.5 months, and median OS is not reached.
Kaplan-Meier estimates of PFS and OS. PFS (A) with 8 events after a median observation time of 26.6 months (range, 3.7 to 41.6) and OS (B) with 2 events. Median PFS is 33.5 months, and median OS is not reached.
Of the 6 patients with PD at the end of follow-up, 4 patients had stopped treatment according to the protocol with a CR, 1 with a PR, and each of them with uMRD (in PB and BM) (Figure 5). The sixth patient stopped treatment at cycle 15 with missing MRD in BM and PB. Three of the patients with CR had no relevant treatment interruptions, and 1 had a dose reduction of ibrutinib due to neutropenia from cycle 9 to cycle 15. The patient with PR who stopped treatment at cycle 15 stopped taking ibrutinib and venetoclax over a 3-month period due to arthralgia. The second patient with PR had a cerebral aspergillosis in cycle 2, and ibrutinib and venetoclax were stopped. Venetoclax was restarted with a highest dose of 200 mg. Progression events occurred between cycles 24 and 34 in 3 patients with CR (nodal progression in all cases) and in the 2 patients with PR (the patient who stopped treatment at cycle 15 developed lymphocytosis, while the other had nodal progression) without further MRD testing at relapse. All 5 of these patients received further therapy following progression. Progression events occurred during follow-up in the other patient with CR (nodal progression and lymphocytosis). As such, this patient reached the end of study, so no more information was available about any further therapy.
Four patients (9.8%) received subsequent treatments: 3 received only 1 subsequent treatment (combination therapy of acalabrutinib, obinutuzumab, and venetoclax [n = 2], venetoclax monotherapy [n = 1]). Both patients receiving the triplet combination responded to the subsequent treatment and achieved a PR. The response assessment for the patient receiving venetoclax monotherapy is unknown. One patient received 2 subsequent treatments with obinutuzumab monotherapy and achieved a PR after the first treatment (response assessment is unknown for the second treatment with obinutuzumab monotherapy).
Safety
AEs were reported in all patients (Table 2); most were low grade (23.8% AEs grade ≥3) and resolved without sequelae (454 of 528, 86.0%). A total of 56 SAEs occurred in 25 patients; the most frequent classified as infections and infestations (16 of 56, 28.6%). Two deaths were reported, 1 classified as concomitant disease (cardiac failure due to aggravated mitral valve insufficiency at cycle 9 in an 86-year-old patient) and 1 as other malignancy (ovarian carcinoma diagnosed at cycle 3 but assessed retrospectively to be preexisting at enrollment). Both deaths were assessed as unrelated to study treatment (supplemental Table 5A).
Overview of safety
| Characteristic | Patients (n = 41) |
|---|---|
| Patients with ≥1 AE, n (%) | 41 (100.0) |
| Total number of AEs, n | 528 |
| Patients with ≥1 AE grade 3-5, n (%) | 32 (78.0) |
| CTC grade for all AEs | |
| 1 | 229 (43.4) |
| 2 | 173 (32.8) |
| 3 | 102 (19.3) |
| 4 | 22 (4.2) |
| 5 | 2 (0.4) |
| Outcome for all AEs, n (%) | |
| Resolved | 454 (86.0) |
| Resolved with sequelae | 8 (1.5) |
| Persisting | 63 (11.9) |
| Death | 2 (0.4) |
| Missing | 1 (0.2) |
| Patients with ≥1 related SAEs, n (%) | 25 (61.0) |
| Total number of SAEs, n | 56 |
| CTC grade for all SAEs, n (%) | |
| 1 | 1 (1.8) |
| 2 | 6 (10.7) |
| 3 | 45 (80.4) |
| 4 | 2 (3.6) |
| 5 | 2 (3.6) |
| Total number of treatment-related SAEs, n (%) | 33 (58.9) |
| Patients who discontinued due to AE, n (%) | 5 (12.2) |
| Total number of AEs leading to permanent discontinuation, n (%) | 5 (0.9) |
| AEs leading to permanent treatment discontinuation by term, max. cycles of treatment (CTCAE Grade)* | |
| Progressive multifocal leukencephalopathy | C9 (2) |
| Ovarian cancer | C3 (5) |
| Cerebral aspergillosis | C12 (4) |
| Atrial fibrilliation | C12 (1) |
| Arthralgia | C15 (1) |
| Patients with ≥1 AE leading to dose modifications, n (%) | 31 (75.6) |
| Total number of AEs leading to dose modifications, n (%) | |
| Reductions | 25 (4.7) |
| Interruptions | 73 (13.8) |
| Category (SOC) with total number of AEs >5%, n (%) | |
| Blood and lymphatic system disorders | 93 (17.6) |
| Gastrointestinal disorders | 91 (17.2) |
| Infections and infestations | 90 (17.0) |
| General disorders and administration site conditions | 39 (7.4) |
| Musculoskeletal and connective tissue disorders | 30 (5.7) |
| Characteristic | Patients (n = 41) |
|---|---|
| Patients with ≥1 AE, n (%) | 41 (100.0) |
| Total number of AEs, n | 528 |
| Patients with ≥1 AE grade 3-5, n (%) | 32 (78.0) |
| CTC grade for all AEs | |
| 1 | 229 (43.4) |
| 2 | 173 (32.8) |
| 3 | 102 (19.3) |
| 4 | 22 (4.2) |
| 5 | 2 (0.4) |
| Outcome for all AEs, n (%) | |
| Resolved | 454 (86.0) |
| Resolved with sequelae | 8 (1.5) |
| Persisting | 63 (11.9) |
| Death | 2 (0.4) |
| Missing | 1 (0.2) |
| Patients with ≥1 related SAEs, n (%) | 25 (61.0) |
| Total number of SAEs, n | 56 |
| CTC grade for all SAEs, n (%) | |
| 1 | 1 (1.8) |
| 2 | 6 (10.7) |
| 3 | 45 (80.4) |
| 4 | 2 (3.6) |
| 5 | 2 (3.6) |
| Total number of treatment-related SAEs, n (%) | 33 (58.9) |
| Patients who discontinued due to AE, n (%) | 5 (12.2) |
| Total number of AEs leading to permanent discontinuation, n (%) | 5 (0.9) |
| AEs leading to permanent treatment discontinuation by term, max. cycles of treatment (CTCAE Grade)* | |
| Progressive multifocal leukencephalopathy | C9 (2) |
| Ovarian cancer | C3 (5) |
| Cerebral aspergillosis | C12 (4) |
| Atrial fibrilliation | C12 (1) |
| Arthralgia | C15 (1) |
| Patients with ≥1 AE leading to dose modifications, n (%) | 31 (75.6) |
| Total number of AEs leading to dose modifications, n (%) | |
| Reductions | 25 (4.7) |
| Interruptions | 73 (13.8) |
| Category (SOC) with total number of AEs >5%, n (%) | |
| Blood and lymphatic system disorders | 93 (17.6) |
| Gastrointestinal disorders | 91 (17.2) |
| Infections and infestations | 90 (17.0) |
| General disorders and administration site conditions | 39 (7.4) |
| Musculoskeletal and connective tissue disorders | 30 (5.7) |
CTC, common terminology criteria; SOC, system organ class; C, cycle.
AEs (and their CTCAE grades in brackets) listed by term and number of maximally reached cycles (C) of treatment, detailed and described in the paragraph “Efficacy.”
Five AEs (0.9%) led to permanent treatment discontinuation in 5 patients (12.2%) (supplemental Table 5B). Overall, 31 patients (75.6%) had AEs leading to dose adjustment; 25 of 528 AEs (4.7%) led to dose reductions and 73 of 528 (13.8%) led to dose interruptions.
No patient experienced clinical TLS, and 4 patients (9.8%) had laboratory TLS grade ≥3 AEs (Table 2).
Most of the AEs reported occurred during the induction phase (329 AEs in all 41 patients) (supplemental Table 1). The most frequent AEs in induction were disorders in blood and lymphatic system and gastrointestinal disorders (both n = 63) and in the consolidation phase disorders in blood and lymphatic system (n = 28), gastrointestinal disorders (n = 24), and infections (n = 21). Most common infections until cycle 15, day 1 were respiratory (n = 22) and urinary tract (n = 8) infections (supplemental Table 5C). Neutropenia contributed to 57.1% (n = 36) of AEs for blood and lymphatic system disorders during induction and 100% (n = 28) in both the consolidation and maintenance phases. All instances of thrombocytopenia occurred during induction, and diarrhea and nausea were only reported in the induction and consolidation phases.
Sixteen patients (39.0%) received a total of 91 G-CSF administrations, ranging from before the start to after the end of study treatment (supplemental Table 3). Median number of administrations per patient was 3.5 (range, 1 to 16) and median duration of administration was 1 day (range 1 to 13). Considering only G-CSF administrations given after the start of study therapy, 16 patients who received G-CSF received it because of ≥1 AE (85 administrations in total), and 3 patients (5 administrations) also received G-CSF due to another reason.
Discussion
The CLL2-GIVe trial of triple combination therapy with obinutuzumab, ibrutinib, and venetoclax in patients with CLL and del(17p) and/or TP53 mutation demonstrated promising efficacy in this high-risk subgroup, meeting its primary endpoint with a CR rate of 58.5% after 14 cycles of therapy with a uMRD rate of 95.8% in PB in patients achieving CR. The safety profile was manageable; although AEs were reported in all patients, the majority were low grade and resolved during continued study treatment.
Del(17p) and/or TP53 mutation is still one of the most significant negative prognostic indicators in the context of BTK or BCL-2 inhibitor treatment.25,29-33 uMRD is associated with durable remission,17,44-46 even in patients with high-risk CLL.16 In the current study, high rates of uMRD were achieved in PB after cycles 9 (uMRD rate 87.8%) and 12 (uMRD rate 85.4%) during triple and double combination therapy, respectively.
Early trials of novel therapies in patients with high-risk CLL were primarily conducted in the relapsed or refractory (R/R) setting. In the M13-982 study, continuous monotherapy with venetoclax in patients with R/R CLL with del(17p)/TP53 mutation demonstrated a 24-month PFS of 54% (95% CI, 45%-62%) and OS of 73% (95% CI, 65%-79%).47 The RESONATE-17 (PCYC 17) trial, where patients with R/R del(17p) CLL or small lymphocytic lymphoma received ibrutinib (420 mg once daily) until PD or unacceptable toxicity, reported 2-year PFS and 2-year OS rates (95% CI) of 63% (54 to 70) and 75% (67 to 81), respectively.48
A very recent ibrutinib monotherapy analysis in high-risk CLL showed in the treatment naïve cohort (n = 27) a PFS of 60% and an OS of 79% after 6 years, indicating that continuous BTKi monotherapy is providing durable remissions in these high-risk patients.49
The pivotal CLL14 trial, which investigated 12 months of venetoclax plus obinutuzumab vs chlorambucil plus obinutuzumab as first-line treatment in patients with CLL with comorbidities demonstrated, in the venetoclax arm, 24 months PFS rate of 64.7% in the del(17p)/TP53 subset (n = 17 patients; median PFS of 29 months; P < .01).13,25 Additionally, after a median follow-up of 39.6 months, patients with TP53 deletion and/or mutation had both inferior PFS and OS vs those without TP53 deletion and/or mutation.50 With a longer follow-up of 4 years, the median PFS for del(17p) in venetoclax/obinutuzumab increased to 47 months. A similar increase in median PFS may be expected with a longer follow-up in CLL2-GIVe. Currently, the 24-month PFS is a more reliable estimate in CLL2-GIVe and, therefore, a better basis for comparison, and suggests a benefit from GIVe in comparison with venetoclax/obinutuzumab in CLL14 in 17p/TP53 patients.26,50 In CLL2-GIVe, both the 2-year PFS rate and 2-year OS rate were 95.1%. Furthermore, 6 progression events had occurred between months 24 and 34 of follow-up, mostly in patients who had finished treatment according to the protocol due to achieving CR/CRi and uMRD at the final restaging at cycle 15. Nevertheless, with a median follow-up of 26.6 months, PFS data are not yet mature enough to draw conclusions regarding the long-term outcome beyond 2 years.
As disease progression occurred more frequently after the end of therapy, periodic MRD testing (eg, MRD >10−4 in blood, every 3 months) may be useful to detect relapse early if CLL is measurable by MRD testing. This is already implemented in an ongoing phase 2 study of acalabrutinib, venetoclax, and obinutuzumab (AVO) for frontline treatment of CLL (n = 37 patients, nearly 27% patients with TP53 aberrant disease). Beyond the end of treatment, MRD was tested every 3 months in blood. In the case of MRD >10−4 in blood, acalabrutinib and venetoclax were restarted. The AVO triplet is highly active, with 86% achieving uMRD in blood in all patients and 80% in patients with TP53 aberrant disease after 15 months of time-limited therapy in a frontline CLL population. After a median follow-up of 27.6 months, no progressions occurred.51,52
Furthermore, more comprehensive and sensitive monitoring might be relevant, for example, the development of circulating tumor DNA-based approaches, as a better tool to detect disease in all compartments.53
Another phase 2 study has also evaluated the combination of obinutuzumab, ibrutinib, and venetoclax for a total of 14 cycles, in patients with treatment-naïve (n = 25) and R/R (n = 25) CLL.14,54 However, only 4 patients in this study had del(17p), indicating the need for further investigations. In contrast to the CLL2-GIVe study, where ORR was 100%, ORR in the other study was 84% for the treatment-naïve subgroup; the most frequent response was PR.54 Notably, PFS and OS were not reached in either cohort, highlighting the promising efficacy for this treatment combination. If continuous BTKi treatment or fixed duration therapy with the potential of retreatment (as described in AVO trial protocol) is more useful, it will be addressed in further larger studies such as in the CLL16 and CLL17 protocols of the German CLL study group.
The overall level of AEs observed was comparable with other studies of double combinations using novel agents.9,13,55-57 Furthermore, types of frequent hematologic and nonhematologic AEs were in line with other studies. Similar numbers of premature discontinuations were reported for patients with high-risk CLL compared with the triplet regimen in treatment-naïve and R/R disease (6 discontinuations and 2 deaths vs 7 discontinuations and 1 death, respectively).54 Of note, the study population enrolled in CLL2-GIVe was mainly fit (CIRS ≤6 in 95.1% of patients), and potential side effects in an older study population remain unclear.
Abnormalities in TP53 are present in up to 50% of patients with R/R disease and only 5% to 15% of treatment-naïve patients.22,25,27,57-59 As such, the enrollment of patients with high-risk CLL according to del(17p)/TP53 mutation is the major limiting factor concerning the general applicability of these results, particularly in regards to investigating the correlation of PFS and TP53 mutation status [where only 2 patients had del(17p) and unmutated TP53].
In conclusion, our results indicate that the CLL2-GIVe regimen is a promising fixed-duration, first-line treatment of patients with TP53-aberrant CLL, with a CR rate of 58.5% and MRD negativity rate of 78.0% in PB at final restaging. The safety profile was manageable. These data provide a rationale to explore this triplet combination not only in high-risk CLL, where the unmet medical need is greatest, but also among all patients with untreated CLL as currently being evaluated within the CLL13/GAIA trial.
Acknowledgments
The authors wish to thank the patients and their families, the investigators at the sites, and the German CLL Study Group study office. We especially want to thank Michaela Grimm and Anke Hallmen of Ulm University Medical Center study office and the team of the German CLL Study Group central office at Cologne University Medical Center for their contribution to the conduct of the study. The trial design was developed by the German CLL Study Group, sponsor of the study was Ulm University Medical Center, and the study was supported by Roche Pharma AG and Janssen Cilag. Sponsor and German CLL Study Group representatives analyzed the data and wrote the first draft of the manuscript. Initial medical editing support was provided by Rachel Dobb and Lynda McEvoy of Ashfield MedComms, an Ashfield Health company.
Authorship
Contribution: Patients were recruited by H.H., S.E., J.v.T., E.T., C.S., J.B., M.F., P.D., M.R., T.I., A.L.I., J.D., S.B., C.U.N., A.-M.F., K.F., H.D., M.H., B.E., and S.S.; E.T., C.S., J.B., and S.S. performed the genetic analyses; first version of the manuscript was written by H.H., S.E., and S.S.; H.H., S.E., J.v.T., B.E., and S.S. designed and managed the clinical trial and interpreted the clinical data; MRD was analyzed by M.R. and M.K.; statistical analyses were performed by S.R., and C.Z.; and all authors critically reviewed and approved the manuscript.
Conflict-of-interest disclosure: J.v.T.: research support: Janssen/Roche; travel grants: Abbvie, Janssen, Roche, Celgene; honoraria: Roche, Janssen, Abbvie, AstraZeneca; advisory boards: Janssen, Roche, Abbvie. H.H.: travel grants: Novartis; honoraria: Abbvie. P.D.: consultancy for AbbVie, AstraZeneca, Bluebird Bio, Gilead, Janssen, Novartis, Riemser, and Roche; speakers bureau for AbbVie, AstraZeneca, Gilead, Novartis, Riemser, and Roche. S.B.: payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events: Roche, AbbVie, Norvatis, Becton Dickinson, Janssen, Astra Zeneca, and Sanofi; grants or contracts from any entity: Janssen Cilag Neuss. A.-M.F.: research funding: Celgene; advisory board: Janssen; and travel grants: AbbVie. C.U.N.: grant research: Abbvie, Janssen, and AstraZeneca; advisory board: Abbvie, Janssen, Gilead, Roche, AstraZeneca, Acerta, and Sunsis; travel reimbursement: Gilead, Roche, Novartis; consultancy: CSL Behring. S.S.: advisory board honoraria, research support, travel support, speaker fees: AbbVie, Amgen, AstraZeneca, Celgene, Gilead, GSK, Hoffmann-La Roche, Janssen, Novartis, and Sunesis. All other authors declare no competing financial interests.
Correspondence: Stephan Stilgenbauer, Ärztlicher Direktor Comprehensive Cancer Center Ulm (CCCU), Leiter Early Clinical Trials Unit (ECTU) CCCU, Leiter Sektion CLL Klinik für Innere Medizin III, Albert-Einstein-Allee 23, 89081 Ulm, Germany; e-mail: stephan.stilgenbauer@uniklinik-ulm.de.
Presented partly as an oral presentation at the 25th European Hematology Association (EHA) Annual Congress: “Unfolding the Future!” 11-21 June, 2020, virtual edition.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked "advertisement" in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal