

Glycosylphosphatidylinositol (GPI) anchors correctly localize 1% to 2% of human proteins to the cell membrane. Although biosynthesis of GPI anchors is commonly inactivated by somatic Phosphatidylinositol glycan anchor biosynthesis class A (PIGA) mutations in hematopoietic precursors in paroxysmal nocturnal hemoglobinuria (PNH), constitutional PIGA mutations are rare and have been implicated in severe impairment of neurologic development. Here we uncover that hypomorphic constitutional PIGA mutations cause a novel subtype of hereditary hemochromatosis (HH) by severely limiting GPI anchorage of hemojuvelin and ceruloplasmin.
HH is a genetic iron overload disease caused by dysfunction of the hepcidin/ferroportin regulatory axis due to mutations in several, mainly liver-expressed genes (high Fe [HFE], transferrin receptor 2 [TfR2], hemojuvelin [HJV], hepcidin [HAMP], or ferroportin [FPN]). The most common HH subtype, with high prevalence in the White population, presents with an adult onset and chronic phenotype and is caused by the HFE p.C282Y mutation. The non-HFE HH subtypes are relatively rare and typically manifest during juvenile age. Hepcidin, the key regulator of systemic iron homeostasis, binds to the iron exporter ferroportin, triggering its degradation to inhibit iron export from duodenal enterocytes, hepatocytes, and macrophages. Inappropriately low hepcidin levels hallmark HH and explain increased intestinal iron absorption, progressive iron accumulation, and damage of parenchymal organs. A severe, juvenile HH subtype is caused by mutations in HJV, a GPI-anchored protein that enhances the bone morphogenetic protein /small mothers against decapentaplegic (SMAD) signaling pathway by functioning as a bone morphogenetic protein coreceptor.1 Some HH-like phenotypes still lack a molecular basis.
In this study, we present the discovery of a novel mutation causing juvenile HH. We investigated a pediatric male patient (patient 1), who, in addition to early-onset epilepsy, severe developmental delay, and intellectual disability, demonstrated early systemic iron overload, meeting the diagnostic criteria for juvenile non-HFE HH.2 Of note are the very high transferrin saturation (TSAT), diminished transferrin, and high serum iron and ferritin levels in the upper normal range. Together with low plasma hepcidin levels, these findings were suggestive of HH.
In this patient, exome sequencing failed to detect known HH-associated mutations and instead identified a constitutional hemizygous missense mutation in the X-linked PIGA. Using next-generation panel sequencing, we could confirm this finding in 2 additional patients (patients 2 and 3), who were initially brought to medical attention because of their neurologic symptoms. Further assessment revealed signs of iron overload (Table 1), which then led to their referral to pediatric hematology. The clinical information of the 3 patients is summarized in Table 1. Supplemental Figure 1 (available on the Blood Web site) displaying the pedigrees of all 3 patients shows a pattern consistent with X-chromosomal inheritance.
Genetic, clinical, and biochemical features in 3 patients with constitutional PIGA deficiency
| Patient 1 | Patient 2 | Patient 3 | |
|---|---|---|---|
| Constitutional PIGA mutation | c.230G>A (p.R77Q) | c.1031T>C (p.L344P) | c.380 C>T (p.S127L) |
| Inheritance | Maternal | Maternal | Maternal |
| Sex | Male | Male | Male |
| Age at time of data collection | 13 y | 7 y | 2 y |
| Degree of developmental delay | Moderate | Severe | Severe |
| Hypotonia | No | Yes | Yes |
| Age at seizure onset | 11 mo | 9 mo | 6 mo |
| Seizure type and severity | Focal and generalized tonic-clonic seizures partially controlled by valproate, stiripentol, and clobazam | Focal and generalized tonic-clonic seizures partially controlled by topiramat, zonisamid and levetiracetam | Focal and atonic seizures, seizure free under the administration of topiramat and lacosamid |
| Brain anomalies | No | Multifocal cortical dysplasia of the right hemisphere (possibly explained by a hemizygous pcdh19 variant) | No |
| Other congenital anomalies | Hypospadia glandis, primary enuresis nocturna | Cryptorchidism, cortical visual impairment, ichthyosis, hyper-/ hypopigmentation on the neck, widely spaced teeth | Somatomegaly |
| Biochemical | |||
| Ferritin (µg/L) | 188 (RR: 7-140) | 96 (RR: 7-140) | 79 (RR: 2-63) |
| Transferrin saturation (%) (RR: 16-45) | 96 | 87 | 70 |
| Transferrin (g/L) (RR: 2.0-3.6) | 1.87 | 1.97 | 2.23 |
| Serum iron (μmol/L) (RR: 14-32) | 45.1 | 43.2 | 39.0 |
| Hepcidin (ng/mL)17 | 2.9 (R:16.58-74.57) | 4.82 (R:6.78-118.86) | 3.1 (R:10.32-115.73) |
| FerriScan (MRI) (mg/g dry tissue) (RR: 0.17-1.8) | 4.0 | 1.8 | NA* |
| Serum transaminases (U/L) | Normal | Normal | Elevated (GOT 57 (RR: < 56); GPT 69 (RR: < 39)) |
| Serum alkaline phosphatase (U/L) | 243 (RR: 118-518) | 223 (RR: 86-315) | 411 (RR: 75-316) |
| Deficiency of GPI-anchored proteins and GPI anchors | ↓ CD48 on a subpopulation of B- and T-lymphocytes (nonsignificant, 0.22%) | ↓ CD157 on a subpopulation of monocytes (nonsignificant; 0.13%) | ↓ CD58 and CD59 on a subpopulation of reticulocytes (not significant due to a very low number of reticulocytes) |
| Patient 1 | Patient 2 | Patient 3 | |
|---|---|---|---|
| Constitutional PIGA mutation | c.230G>A (p.R77Q) | c.1031T>C (p.L344P) | c.380 C>T (p.S127L) |
| Inheritance | Maternal | Maternal | Maternal |
| Sex | Male | Male | Male |
| Age at time of data collection | 13 y | 7 y | 2 y |
| Degree of developmental delay | Moderate | Severe | Severe |
| Hypotonia | No | Yes | Yes |
| Age at seizure onset | 11 mo | 9 mo | 6 mo |
| Seizure type and severity | Focal and generalized tonic-clonic seizures partially controlled by valproate, stiripentol, and clobazam | Focal and generalized tonic-clonic seizures partially controlled by topiramat, zonisamid and levetiracetam | Focal and atonic seizures, seizure free under the administration of topiramat and lacosamid |
| Brain anomalies | No | Multifocal cortical dysplasia of the right hemisphere (possibly explained by a hemizygous pcdh19 variant) | No |
| Other congenital anomalies | Hypospadia glandis, primary enuresis nocturna | Cryptorchidism, cortical visual impairment, ichthyosis, hyper-/ hypopigmentation on the neck, widely spaced teeth | Somatomegaly |
| Biochemical | |||
| Ferritin (µg/L) | 188 (RR: 7-140) | 96 (RR: 7-140) | 79 (RR: 2-63) |
| Transferrin saturation (%) (RR: 16-45) | 96 | 87 | 70 |
| Transferrin (g/L) (RR: 2.0-3.6) | 1.87 | 1.97 | 2.23 |
| Serum iron (μmol/L) (RR: 14-32) | 45.1 | 43.2 | 39.0 |
| Hepcidin (ng/mL)17 | 2.9 (R:16.58-74.57) | 4.82 (R:6.78-118.86) | 3.1 (R:10.32-115.73) |
| FerriScan (MRI) (mg/g dry tissue) (RR: 0.17-1.8) | 4.0 | 1.8 | NA* |
| Serum transaminases (U/L) | Normal | Normal | Elevated (GOT 57 (RR: < 56); GPT 69 (RR: < 39)) |
| Serum alkaline phosphatase (U/L) | 243 (RR: 118-518) | 223 (RR: 86-315) | 411 (RR: 75-316) |
| Deficiency of GPI-anchored proteins and GPI anchors | ↓ CD48 on a subpopulation of B- and T-lymphocytes (nonsignificant, 0.22%) | ↓ CD157 on a subpopulation of monocytes (nonsignificant; 0.13%) | ↓ CD58 and CD59 on a subpopulation of reticulocytes (not significant due to a very low number of reticulocytes) |
c.230G>A (p.R77Q): ClinVar18 VCV000810512, likely pathogenic; PolyPhen-219 prediction, probably damaging; c.1031T>C (p.L344P): ClinVar VCV000444793, uncertain significance; PolyPhen-2 prediction: probably damaging; c.380 C>T (p.S127L): ClinVar, not annotated; PolyPhen-2 prediction, likely damaging.
NA, not available; R, range; RR, reference range.
Not indicated at the age of 2 y because sedation would be necessary.
PIGA catalyzes the first step of GPI anchor biosynthesis, a process that is important for the dynamics and cell membrane attachment of approximately 150 human proteins.3 Somatic PIGA mutations have been described in patients with PNH (Online Mendelian Inheritance in Man [OMIM] 300818), a clonal hematopoietic stem cell disorder hallmarked by severe deficiency of GPI anchors that manifests with hemolytic anemia, thrombosis, and bone marrow failure.4 Constitutional PIGA mutations causing PIGA deficiency are very rare and show a wide phenotypic spectrum including epileptic seizures, profound developmental delay, intellectual disability, and multiple congenital malformations.5 In line with previously described children with constitutional PIGA mutations, our patients do not show clinical or biochemical signs of PNH (details in supplemental Table 1) and do not meet the flow cytometric criteria for this condition. This is likely explained by residual activity of the hypomorphic mutations identified here, which result in a less severe reduced function than the acquired somatic null mutations causing PNH. This interpretation is consistent with the observation that not all blood cell lineages show a reduction of GPI-anchored proteins (Table 1).
PIGA deficiency has, thus far, not been known to cause HH hallmarked by hepcidin deficiency. However, HJV is a GPI-anchored protein,6 and we and others7 hypothesized that iron overload could be caused by a failure to attach GPI anchors to HJV and a subsequent inability to appropriately induce hepcidin expression by hepatocytes.
To test this hypothesis, we performed Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas12a-mediated knockout (KO)8 of PIGA in Hep3B liver cells and analyzed their capacity to control hepcidin expression. The PIGA gene is localized on the X chromosome, and therefore, we selected Hep3B cells that are derived from a male patient with hepatocellular carcinoma and consequently only contain 1 X chromosome.9 We inserted a polymerase chain reaction cassette (Figure 1A) containing 2 selection markers, puromycin and iScarlet, into exon 2 of the gene, disrupting the PIGA open reading frame in PIGA KO clones. Reference cells were either clones that underwent the same CRISPR/Cas12a-mediated procedure, but in which PIGA was not ablated (from now on referred to as PIGA wild-type [WT] clones) or parental Hep3B cells (Figure 1B-C). To demonstrate efficient and specific inhibition of GPI anchoring of proteins in PIGA KO cells, we performed flow cytometry analysis for cell surface expression of the GPI-anchored protein CD59, a diagnostic marker analyzed in patients with somatic PIGA mutations and PNH.4 As shown in Figure 1D, CD59 is only detectable in PIGA WT but not in PIGA KO cells. We next investigated the functionality of HJV in PIGA KO cells. Because specific, high-affinity antibodies against HJV are not available, we overexpressed a Flag-tagged version of the protein and analyzed HJV surface expression by flow cytometry analysis using an anti-Flag antibody. Our data show an almost complete absence of cells with HJV surface expression in PIGA KO clones (Figure 1E), whereas HJV is readily detectable on the surface of PIGA WT cells. Consistently, only WT cells show the expected upregulation of hepcidin in response to HJV overexpression,10 whereas PIGA KO cells fail to do so. Importantly, simultaneous transfection of HJV with a PIGA expression construct rescues hepcidin expression in PIGA KO cells (Figure 1F) and raises other markers of SMAD signaling, such as SMAD6 and SMAD7 expression (supplemental Figure 2). We next coexpressed the PIGA mutations identified in our patients (Table 1) or those described in patients reported previously with severe neurologic phenotypes.7,11 Importantly, PIGA KO cells show significantly lower hepcidin mRNA levels on HJV overexpression with all PIGA mutants (L344P, R77Q, L110del, and R412*) in comparison with the PIGA WT construct (Figure 1G). These data directly demonstrate the functional deficiency of the patients’ PIGA alleles and offer a molecular explanation for the increased TSAT and other iron overload parameters observed in our patients with the L344P and R77Q mutations. Of note, hepcidin mRNA levels are particularly low upon overexpression of the PIGA L110del and R412* mutations, conceivably accounting for the iron overload phenotype previously observed in the patients described by Swoboda et al7 with the PIGA L110del mutation. In comparison with patients with constitutional PIGA mutations displaying a more severe phenotype (exemplified by the R412* stop-gain mutation12), the data of the rescue experiments (Figure 1G) and the less severe clinical phenotype of the affected patients indicate that the missense mutations of our patients retain a higher residual function, thus likely explaining the milder neurologic phenotype allowing for sufficiently long survival to develop the iron overload phenotype. Moreover, our findings suggest that germline hypomorphic mutations in additional critical genes for the biosynthesis of GPI anchors may also cause iron overload over time, as has been recently demonstrated by Tremblay-Laganière et al.13 Interestingly, PIGA KO clones not only lack HJV but also show reduced levels of ceruloplasmin (CP), a ferroxidase required for efficient cellular iron export. Although the failure to express appropriate levels of HJV explains low hepcidin levels and iron accumulation, iron overload may be further aggravated by reduced CP protein expression (Figure 1H). Importantly, a GPI-anchored form of this enzyme is also expressed by astrocytes in the mammalian central nervous system.14 The neurologic phenotype in patients with inactivating PIGA mutations is generally attributed to the impaired attachment of GPI-anchored proteins involved in brain development to the cell membrane. These proteins include the HJV homologs repulsive guidance molecule a (RGMa) and RGMb/DRG11-responsive axonal guidance and outgrowth of neurite (DRAGON), which mediate axon guidance and growth and the formation of neuronal networks and are expressed in the central nervous system and other tissues.15 Decreased CP levels and cellular iron overload may further aggravate the neurologic symptoms of patients with constitutional PIGA mutations (eg, hypotonia and movement disorders), reminiscent of observations in patients with aceruloplasminemia.16 In addition, these patients are expected to have high levels of non–transferrin-bound iron (NTBI), the free toxic form of iron, because of an excessively high TSAT, which will further damage various cell types including neuronal cells.
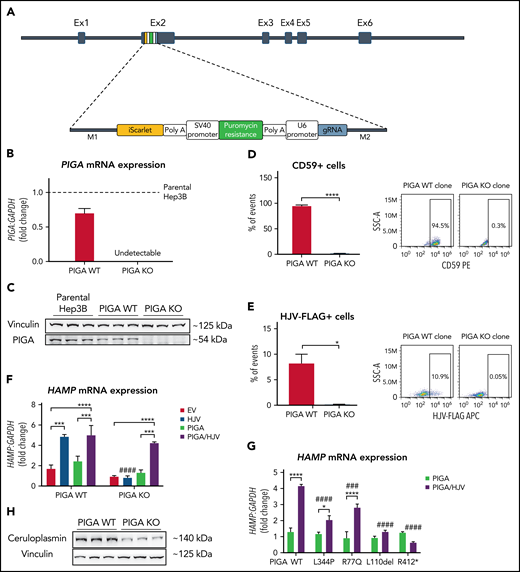
PIGA deficiency impairs hepcidin (HAMP) upregulation upon HJV overexpression. (A) Schematic representation of the PCR cassette integration in exon (Ex) 2 of PIGA. (B) mRNA levels of PIGA in PIGA WT and PIGA KO cells, normalized to parental Hep3B cells (dashed line). PIGA mRNA expression was normalized to the housekeeping gene GAPDH. (C) Western blot analysis of PIGA in parental Hep3B, PIGA WT, and PIGA KO cells. Vinculin was used as a loading control. (D) Fluorescence-activated cell sorter (FACS) analysis representing the percentage of CD59 hycoerythrin-positive cells in PIGA WT and PIGA KO clones. (E) FACS analysis of the percentage of HJV-Flag allophycocyanin-positive cells in PIGA WT and PIGA KO cells upon HJV-Flag overexpression. In panels D and E, cells were previously gated for the exclusive inclusion of singlets (FSC-A vs FSC-H) and exclusion of dead cells (7-AAD positive). (F) Hepcidin mRNA levels in PIGA WT and PIGA KO cells after overexpression of HJV, PIGA, or HJV and PIGA vectors simultaneously. EV, empty vector. (G) mRNA levels of Hepcidin in PIGA KO cells after overexpression of PIGAWT, PIGAL344P, PIGAR77Q, PIGAL110del, and PIGAR412* mutants together with HJV. Hepcidin mRNA expression was normalized to the housekeeping gene GAPDH. (H) Western blot analysis of ceruloplasmin (CP) in PIGA WT and PIGA KO cells. Vinculin was used as a loading control. Student t test/2-way analysis of variance: */#P < .05, **/##P < .01, ***/###P < .001, ****/####P < .0001; *comparisons inside each group; #relative to comparisons to the corresponding WT counterpart.
PIGA deficiency impairs hepcidin (HAMP) upregulation upon HJV overexpression. (A) Schematic representation of the PCR cassette integration in exon (Ex) 2 of PIGA. (B) mRNA levels of PIGA in PIGA WT and PIGA KO cells, normalized to parental Hep3B cells (dashed line). PIGA mRNA expression was normalized to the housekeeping gene GAPDH. (C) Western blot analysis of PIGA in parental Hep3B, PIGA WT, and PIGA KO cells. Vinculin was used as a loading control. (D) Fluorescence-activated cell sorter (FACS) analysis representing the percentage of CD59 hycoerythrin-positive cells in PIGA WT and PIGA KO clones. (E) FACS analysis of the percentage of HJV-Flag allophycocyanin-positive cells in PIGA WT and PIGA KO cells upon HJV-Flag overexpression. In panels D and E, cells were previously gated for the exclusive inclusion of singlets (FSC-A vs FSC-H) and exclusion of dead cells (7-AAD positive). (F) Hepcidin mRNA levels in PIGA WT and PIGA KO cells after overexpression of HJV, PIGA, or HJV and PIGA vectors simultaneously. EV, empty vector. (G) mRNA levels of Hepcidin in PIGA KO cells after overexpression of PIGAWT, PIGAL344P, PIGAR77Q, PIGAL110del, and PIGAR412* mutants together with HJV. Hepcidin mRNA expression was normalized to the housekeeping gene GAPDH. (H) Western blot analysis of ceruloplasmin (CP) in PIGA WT and PIGA KO cells. Vinculin was used as a loading control. Student t test/2-way analysis of variance: */#P < .05, **/##P < .01, ***/###P < .001, ****/####P < .0001; *comparisons inside each group; #relative to comparisons to the corresponding WT counterpart.
Taken together our results connect neurologic deficits and iron overload in a novel way and uncover a new form of HH in patients with neurologic deficits and its likely molecular basis, showing that the functionality of 2 GPI-anchored proteins involved in maintaining iron homeostasis, HJV and CP, is impaired by PIGA mutations. These findings call for clinical assessment and treatment of potential iron overload in patients with constitutional PIGA mutations and long-term survival.
Acknowledgments
The authors thank the participants and families involved in the study; Richard Sparla for technical assistance; EMBL GeneCore for sequencing support; and Ana Rita da Silva for technical and scientific advice in the beginning of the project.
M.U.M. acknowledges funding from the Deutsche Forschungsgemeinschaft (DFG) (SFB1036 and SFB1118) and from the Federal Ministry of Education and Research (NephrESA Nr 031L0191C and DZL TLRC-H). M.U.M. and S.A. acknowledge DFG funding for the FerrOs-FOR5146 grant. M.U.M. and A.E.K. acknowledge funding from the Dietmar Hopp Stiftung. O.M. and S.A. received support from the European Hematology Association (Junior [RG66] and Advanced Research Grants, respectively). L.M. is the recipient of a Heidelberg Research Center for Molecular Medicine, Heidelberg junior career fellowship. O.M. is supported by an Olympia Morata-Programm fellowship given by the Medical Faculty of the University of Heidelberg.
Authorship
Contribution: L.M., O.M., S.C., P.F., S.A., B.H., H.S., M.L., N.G., A.E.K., and M.U.M. designed research; L.M., O.M., S.C., P.F., S.A., B.H., H.S., M.L., and N.H.-M. performed experiments; L.M., O.M., S.C., J.K., T.B., B.H., H.S., P.R.-P., T.R., N.H-M., M.W.H., A.E.K., and M.U.M. collected and/or analyzed data; L.M., O.M., N.G., M.W.H., A.E.K., and M.U.M. wrote the manuscript; and all authors revised the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Martina Muckenthaler, Im Neuenheimer Feld 350, 69120 Heidelberg, Germany; e-mail: martina.muckenthaler@med.uni-heidelberg.de.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal