

In this issue of Blood, Kondreddy et al1 reveal a novel endothelial cellular pathway that directly promotes inflammatory-mediated venous thrombogenesis, for which an exogenous nonanticoagulant inhibitor is effective. This pathway involves the Gab2 protein (Grb-associated binder to adaptor signaling protein), which is downstream from interleukin β-1 (IL-β1) signaling and through which the pathway confers MALT1 activation as well as CARAMA-3 phosphorylation. This mediates intracellular activation of Rho and the NF-κB–mediated processes that drive thrombogenesis (see figure).
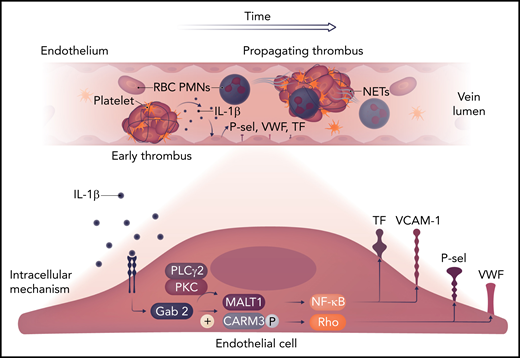
The early venous thrombotic events are shown, with polymorphonuclear neutrophils (PMNs) as likely sources of IL-1β. This stimulates the endothelial Gab2 pathway, which, via MALT1 and CARAMA3 (CARM3), activates NF-κB and Rho signaling, with tissue factor (TF) and von Willebrand factor (VWF) release, as well as P-selectin (P-sel) and VCAM-1 expression. These factors act to amplify the thromboinflammatory process. NET, neutrophil extracellular trap; PKC, protein kinase C; PLC, phospholipase C; RBC, red blood cell. Professional illustration by Somersault18:24.
The early venous thrombotic events are shown, with polymorphonuclear neutrophils (PMNs) as likely sources of IL-1β. This stimulates the endothelial Gab2 pathway, which, via MALT1 and CARAMA3 (CARM3), activates NF-κB and Rho signaling, with tissue factor (TF) and von Willebrand factor (VWF) release, as well as P-selectin (P-sel) and VCAM-1 expression. These factors act to amplify the thromboinflammatory process. NET, neutrophil extracellular trap; PKC, protein kinase C; PLC, phospholipase C; RBC, red blood cell. Professional illustration by Somersault18:24.
Deep vein thrombosis (DVT), along with its sequelae of postthrombotic syndrome and pulmonary embolism (PE), is an extremely common cause of vascular morbidity and mortality. The cornerstone of DVT prophylaxis and treatment is anticoagulation, which affects both hemostasis and thrombosis. Recently, the advent of direct oral anticoagulants against factor X (FX) or FII has made anticoagulation in patients with DVT or PE safer, but there are still significant bleeding risks.2 Moreover, even with the advent of FXIa inhibitors, which mainly affect thrombosis but not hemostasis, these agents have not been evaluated for treatment. The notion that DVT is primarily a thromboinflammatory-mediated event, supported by multiple basic and translational investigations, suggests targets that may ameliorate pathogenic thrombosis without impairing hemostasis.
Various nonhemostatic targets have been proposed, including leukocytes, cell adhesion molecules, neutrophil extracellular traps, and hemostatic targets, such as TF, VWF, and other early mediators of venous thrombogenesis.3 The associated etiologic factors of DVT codified by Virchow’s triad are still relevant and include blood hypercoagulability, blood stasis, and endothelial (or vein wall) injury. It is the latter factor that has been understudied with regard to its role in VT. The endothelium is the primary interface for communication between the thrombus and vein wall as well as between the thrombus and luminal occlusion that results after a thrombotic stimulus. The endothelium confers the natural anticoagulant and antithrombotic milieu that prevent pathologic thrombosis with a high degree of fidelity. However, the endothelial phenotype may convert to a prothrombotic state with various systemic or local insults and is likely central to thrombotic events.4 Experimentally, venous stasis causes endothelial gaps and denudation, with rapid TF expression.5
The strengths of this report are several; first, the well-controlled and elegantly performed in vitro experiments with small interfering RNA and direct pathway inhibitors convincingly demonstrate how Gab2 and downstream processes promote an endothelial prothrombotic state. This is reflected by relevant prothrombotic tissue readouts such as VWF release, P-selectin expression, polymorphonuclear neutrophil adhesion, and TF and VCAM-1 expression.3 Another strength of this report is that 2 models of venous thrombogenesis were used. This is critical, because in human DVT, both complete stasis and partial status are often present, as shown by duplex ultrasound. The stenotic flow–restricted model,6 which is the most commonly used in investigations of venous thrombogenesis, is a neutrophil extracellular trap–dependent model. The investigators also replicated their results using a complete stasis model, with an occlusive thrombus, reflective of a fully obstructed venous segment. To translate the in vitro findings, the investigators used an exogenous MALT1 pathway inhibitor, mepazine. This agent inhibited thrombogenesis (both size and incidence) in both models of VT in the mouse. Interestingly, this agent seemed to significantly decrease the leukocyte content of the thrombus at 2 days, including both neutrophils and monocytes.
There are several aspects that are important but were not addressed in the work presented. First, as a limitation of any in vitro work, the venous endothelium is normally exposed to certain levels of shear stress and stretch in vivo. These biomechanical influences are not replicated with human umbilical vein endothelial cells but are likely relevant. Second, it is unclear whether mepazine directly affects platelet function or release of IL-1β7 or if it only affects the downstream in vivo endothelial cell pathway. It would be easy enough to test if mepazine directly affects platelet reactivity, including a simple test such as tail bleeding time or other platelet activation assays. Third, it would be interesting to know if mepazine affects the monocyte/macrophage phenotype, because recent studies have shown that promoting a noninflammatory phenotype may significantly inhibit thrombogenesis and promote VT resolution, with a similar magnitude of effect as mepazine.8,9
Lastly, it will be important to determine if Gab2 pathway inhibition promotes later VT resolution and associated vein wall injury. Although mepazine may be an ideal agent theoretically to inhibit the formation of pathologic DVT, whether it would be effective as treatment after 48 hours of thrombus formation is not answered in this report. It would be easy to perform experiments evaluating mepazine and would be translationally useful. Along these lines, it would be important to determine if mepazine positively promotes the monocyte phenotype to a prohealing cell, because this may have the benefit of hastening vein wall healing and decreasing fibrosis. Alternatively, altering the endothelial phenotype may also inhibit fibrosis.4
Taken together, the Gab2 pathway is an intriguing endothelial cell target, with potential human translation. Confirmation of these results by other investigators would be appropriate, and examination of the later effects of mepazine as an MALT1 inhibitor on the later cellular and VT resolution functions is a logical next step; definition of any off-target effects is essential as well. The authors should be applauded for considering the endothelium, a part of Virchow’s triad, which has long been neglected, in preventing pathologic VT.
Conflict-of-interest disclosure: P.H. declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal